
生工技术 | 使用MEGA4.1软件构件进化树
许多做了测序的小伙伴需要做进化树辅助数据库比对结果,进化树是用来描述物种之间进化关系的树枝状图形。
我们将演示如何用MEGA4.1软件构件一棵进化树:
序列准备
以研究细菌进化关系为例,首先将测序得到的16s序列BLAST比对下:
在比对结果中,选取需要的,得到这些比对结果结果的FASTA序列:

新建TXT文件,命名某某进化树,里面的序列用>和菌的名称+登录号命名,序列依次粘贴,当然我们还要选择差距大些的才能分支比较明显,比如选择种属都不相同的序列,这样做出的树才会有差异性。
做好的TXT格式如下:
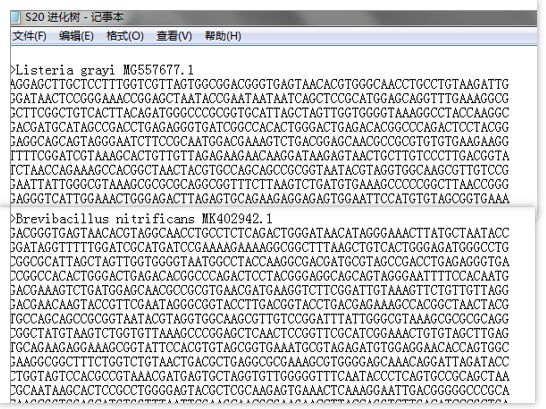
当然,该文本文件可以添加任何需要构建进化树的序列。
MGEA4.1软件对序列进行排布
打开MGEA4.1软件:
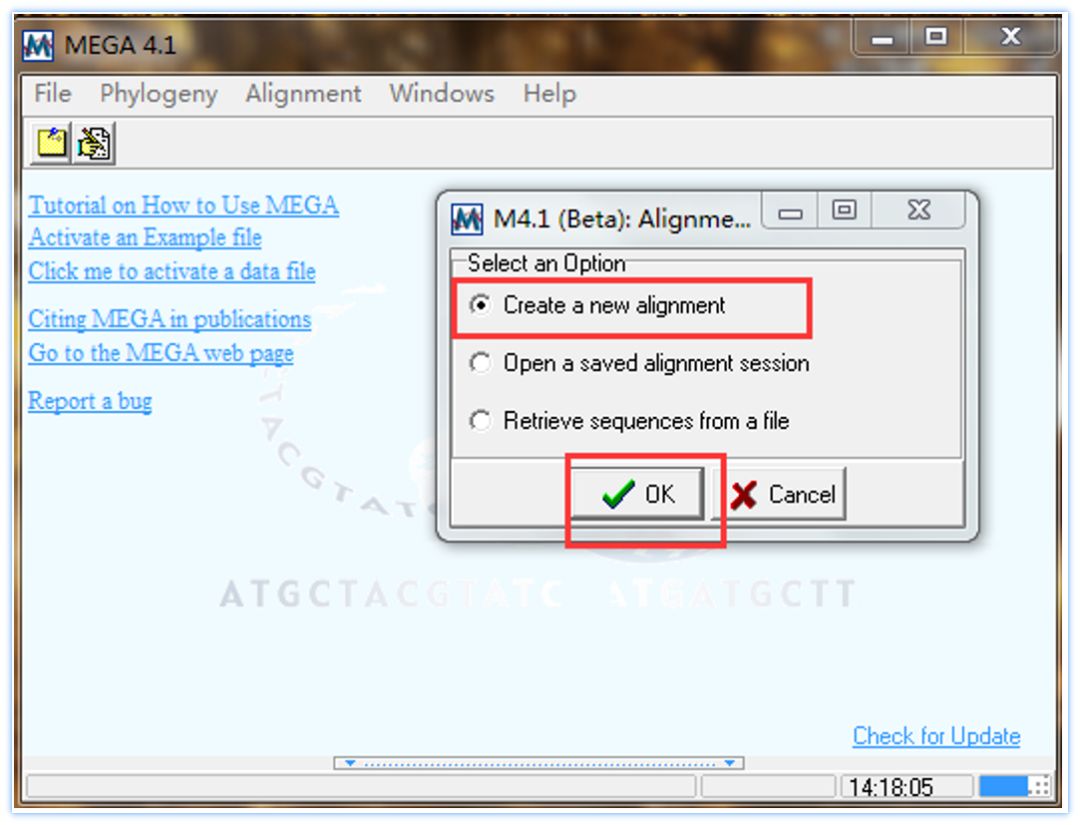
点击Alignment里面的Alignment Explorer/CLUSTAL出现如下界面,选择Create a new alignment,单击OK。
Create a new alignment :是在没有任何比对的时候使用。
Open a saved alignment session:使用它可以打开一个我们已经比对好的序列文件;
Retieve a sequence from a file :这种情况同第一种情况相似,只是不用选择是DNA 还是蛋白质序列比对,打开后的界面都是一样的。
我们比对的是DNA序列所以选择Yes:
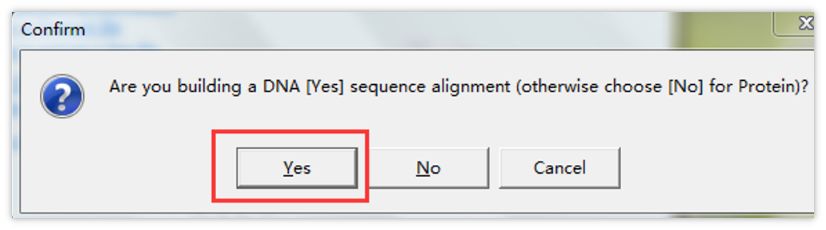
选择红色框添加序列:
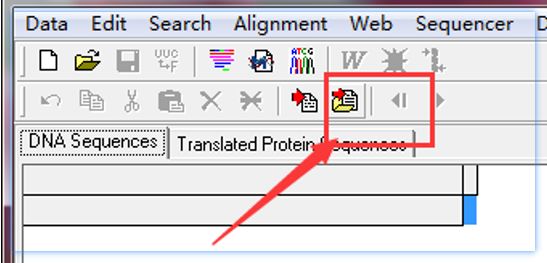
添加序列之后如下,选择Align by Clustal W:
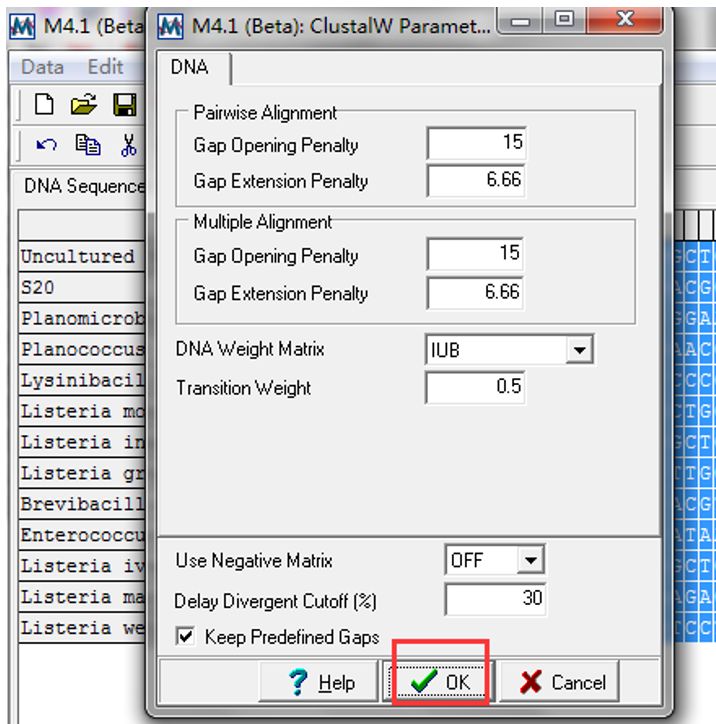
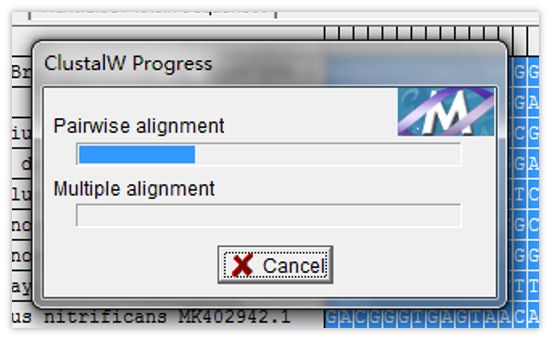
ClustalW是一个多重比对软件,两两对齐和多重对齐进度如下:
选择Data里面的Exit AlnExplorer跳出将对齐界面保存到文件夹中吗,选择YES。
命名、保存后又跳出要不要保存MEGA文件,也选择YES。
MGEA4.1软件对排布序列构建进化树
用MEGA打开刚保存的比对过的文件,选择YES,
然后我们看MEGA软件的整个界面发生了变化,多了几个选项:
我们选择Phylogeny下面的Bootstrap Test of phyloseny里面的邻接法Neighbor-Joining,选择参数如下:

将“bootstrap”自展值设定为重复1000次。
计算模式选择“Model”选择“Kimura 2-parameter”,这是常规建树的选择模式。
加强建树的精度方面“Gamma parameter”,参数可以根据自己的要求设定。在这里采用缺省值“1.0”。并点击 “computer” 进行计算。
建树结果如下:
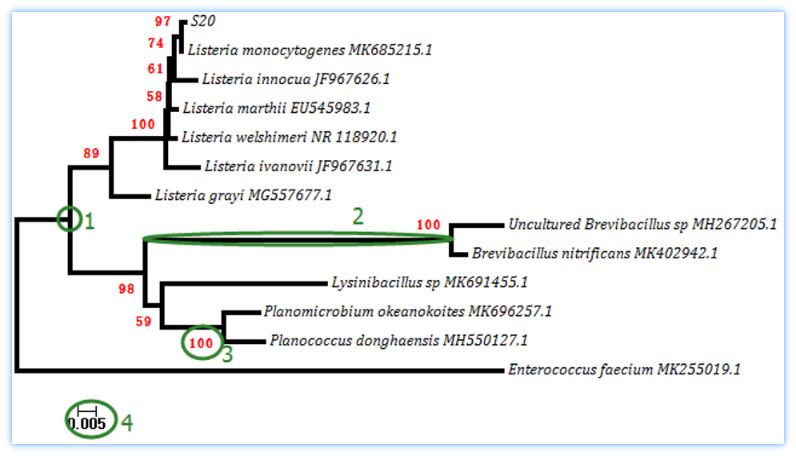
如下解释下进化树上的各数值:
1. 节点:表示一个分支单元.
2. 进化分支长度(遗传距离):分支长度越长表示该分支对应物种的变化越大.
3. Bootstrap值:该值的范围是1%-100%,值越大该节点置信度越高。
4. 距离标尺:下部的数字指该长度的分支代表基因组的遗传变异度。
点击“caption”,将显示本次进化树的所有参数内容。
更多MGEA菜单项解释可以参考如下链接,文章写的不错,分享一下:https://liucheng.name/612/
