
生工技术 | 关于转录组测序的灵魂问答
前言:
对于大多数转录组测序相关初学者来说,会遇到各种转录组实验、分析相关的问题,生工生物高通量测序部整理了被问及最多的20个问题集锦,希望可以帮助到一部分初学者。
什么是转录组测序?
广义的转录组是指在某一状态下,细胞/组织转录出的mRNA、ncRNA(包括lncRNA、circRNA、miRNA、rRNA)等产物的集合,狭义上仅指转录出的mRMA。转录组具有时间特异性、组织特异性、空间特异性等特点。利用高通量测序技术,对上述ncRNA或者mRNA进行大规模测序,以确定其序列及表达量,即是转录组测序。
转录组测序的主要目的是什么?
转录组测序主要适用于筛选相同物种在不同的生长阶段或者发育过程(时间顺序)中差异表达的RNA;或者同一个物种在不同的药物、病原菌、物理化学处理等逆境胁迫处理条件(实验组vs对照组)下差异表达的RNA。
转录组测序如何做生物学重复?怎么取样?
为了结果的科学严谨性,强烈建议做生物学重复,至少3个生物学重复,以排除个体差异导致的假阳性/阴性结果。
生物学重复取样要求:
植物取样:同一片试验田,同一长势,外部形态特征相同;
动物取样:同一遗传背景,同一饲养条件,同一年龄,性别相同,外部形态特征相同;
混合取样:保证混合样品处理方式相同,处于同一发育阶段,个体外部形态特征相同,实质是混合群体重复。
注意:混合取样是针对一个小苗/组织个体抽提的RNA无法达到送样要求,而需要混合多个个体抽提RNA的情况。具体建议如下:2~3个单株混在一起当作一个样品,再取另外与前面所取的单株生长差异不大的2~3个单株混在一起当作前一个样品的生物学重复。
样本检测时需要达到什么要求才认为样本合格?
样本检测主要关注的指标为RNA总量、RIN值、OD260/280以及28S/18S(原核生物为23S/16S)。其中RIN值及28S/18S是评估RNA完整性的主要指标,RIN值越高越好(一般要求RIN值大于7),28S/18S越接近2表明完整性越好。但对于一些特殊样品,比如某些昆虫和水产动物,RNA有隐裂现象,导致没有28S条带,就不能参考RIN值,一般只要18S前基线平稳可认为RNA完整度较高。浓度一般要求大于100ng/ul,总量大于1~2ug。OD260/280=2.0左右表明RNA纯度较高。
普通转录文库与链特异性文库的区别?
与普通文库相比,链特异性文库是在cDNA二链合成时用dUTP代替dTTP,PCR前采用UNG酶消化掉含dUTP的DNA单链,与Flowcell结合时只保留单一链模板,具体流程见下图:
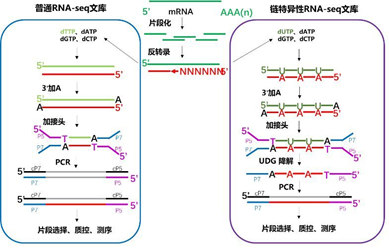
链特异性文库能够保留链的方向性,区分reads是来自于哪条链,减少比对错误,使得基因表达定量更精确、可变剪切检测更准确、非编码转录本的检出率增高和新转录本预测更真实等。
原核生物与真核生物在进行转录组测序文库构建时有什么区别?
原核生物并不像真核生物mRNA具有polyA的结构,因此,无法直接利用 oligo(dT) 磁珠将mRNA 纯化出来。
如果拿total RNA直接进行反转录并测序,那么测序的数据利用率会比较低,因为大部分的序列都来自rRNA。目前,提高原核生物中mRNA的比例,较为主要的方式是采用试剂盒去除total RNA中的rRNA,然后将剩余的RNA用于建库测序。
进行microRNA测序对于组织样品提取总RNA有什么特殊要求?
如果要进行microRNA测序,建议采用乙醇/异丙醇沉淀的方式提取总RNA或者采用专用的small RNA(小于200bp)的提取试剂盒提取。不能使用硅胶膜离心柱试剂盒,也不要使用LiCl沉淀,以免丢失小片段RNA。
转录组测序推荐的测序数据量是多少?
转录组测序所需数据量与所研究物种的基因组大小有关,基因组越大,则所需数据量越大。按照我们的经验来说:常规物种一般建议6G数据即可。一些基因组比较复杂的样品例如小麦等,可以适当增加至8~10G数据量。
做lncRNA测序前需考虑什么问题?
在做lncRNA测序之前,除了生物学重复,样品设置等基本问题需要考虑外,有两个关键点是我们必须要注意的:
1)实验建库环节
是否有此物种rRNA去除的试剂盒(并不是所有的物种都已有成熟的rRNA去除试剂盒);
2)信息分析环节
是否有研究物种组装版本较好的参考基因组(建议染色体组装水平)及注释信息完整的注释文件。
如何确定研究物种有无参考基因组?
根据研究物种的拉丁文名,可在:
Ensembl(http://asia.ensembl.org/index.html)
JGI (http://genome.jgi-psf.org)
或NCBI (http://www.ncbi.nlm.nih.gov/)
等数据库中搜索是否有该物种的基因组信息,也可在其他专门介绍某种物种的数据库网站寻找参考基因组。
测序数据质控指标有哪些?
测序的质控标准主要包括以下指标:
-
Cleanreads占Rawreads的比例情况:
CleanReads即高质量可用reads,Cleanreads比例=Rawreads-低质量reads-含Nreads-接头污染reads
-
测序碱基分布有无AT、GC分离现象:
根据碱基互补原则,A和T的比例应该接近,C和G的比例也应该接近
-
Reads比对比率情况:
理论上,来自成熟mRNA的reads,应该比对到外显子区(Exon),但是存在一些原因导致一部分reads比对到内含子区(Intron)和基因间区(Intergentic);如果参考基因组与测序样品品种存在差异,则也会导致比对到外显子区的reads偏少
-
质量值大于30(Q30)的碱基在cleandata中的占比情况
-
测序数据在全基因组覆盖范围是否均一等
转录组测序可以同时测到mRNA、lncRNA、micRNA以及circRNA么?
狭义的转录组测序只能测到mRNA。
但是全转录组测序通过构建两个测序文库(一是小RNA测序文库、二是lncRNA测序文库)则可以测到以上4种RNA的。
差异基因的筛选标准是什么?是固定不变的吗?
默认差异基因的筛选标准是:表达量差异倍数大于2倍,即foldchange值大于2,且p值或者FDR值小于0.05。不过该标准不是一成不变的,可以要根据实际的情况进行参数调整。
差异基因数目多少比较合理?
不同物种,不同处理,不同组织,不同筛选条件,差异基因数目是不同的,从几十个到几千个都有可能。
但是如果差异基因数目是个位数或者上万个时,就需要和生信分析人员沟通确认是否有问题。
什么时候选择有参考基因组分析,什么时候选择无参分析?
按照有参或者无参进行转录组分析,取决于基因组的质量、所研究物种与参考基因组的比对率。
具体如下:
-
若参考基因组质量较差,则可以选择按照无参转录组分析策略进行分析;
-
若所研究物种与参考基因组比对效率比较低,则需要按照无参转录组分析策略进行分析。一般来说,与参考基因组的比对率在70%以上时,该基因组可以满足后续的分析需求。当比对率低于60%时,需要考虑换参考基因组或者按照无参转录组分析策略进行分析。
如何挑选基因做qPCR验证?
目前,没有统一固定挑选的标准,需要研究人员根据自己的研究需要进行选择。
几个挑选基因的原则如下:
a.一般情况下,需要验证的基因的数目建议不低于20个;
b.样本间表达差异倍数大(Foldchange值大于2);
c.基因表达量较高(至少在一个样品中的表达量高);
d.同时包含上调基因及下调基因;
e.能设计出好的实时定量引物。
qPCR验证不一致的原因是什么?
使用qPCR验证的RNA-Seq定量结果,由于两种技术本身的差异及表达量计算原理的差异,出现不一致的情况属于正常现象,通常一致性大于80%以上。
所以建议关注两种基因表达的检测结果变化趋势总体是不是一致,具体表达量的值及差异倍数数据作为参考。
如果变化趋势不一致,可能的原因有:
1)要保证测序时所用样品同qPCR实验中所用是同一批材料且处理条件一致;
2)尽量选取表达量高的基因进行验证,同时差异倍数在5~10倍的基因更合适;
3)考虑qPCR实验的实验方案、引物序列及原始结果。比如设计探针是否考虑多转录本情况,转录组测序是对转录本。如该基因对应多个转录本,则可能有偏差;
4)该基因是否存在新的可变剪切。
差异基因太多,注释信息太杂乱,怎么挑选目标基因?
建议:
1) 可以根据KEGG和GO富集分析结果,挑选富集程度较高的代谢通路和GO terms,进而查看相关的差异基因;
2) 对不同的差异组合进行维恩图分析,挑选共有或者特有的差异基因作为后续的研究对象;
3) 根据前人的文献报道,挑选相关差异基因,不要局限在自己研究的物种上。
CDS和ORF的区别是什么?
CDS (Coding Sequence,编码序列)是编码蛋白质的一段序列,ORF (Open Reading Frame,开放式阅读框)是从起始密码子到终止密码子的一段序列。
不是所有的读码框都能表达出蛋白质,也就是说CDS一定是ORF,但ORF不一定是CDS,一般ORF只是理论上的一个编码区,CDS则比较接近实际情况。在预测CDS的时候是先跟数据库比对,比对上的直接提取CDS序列,比对不上的再用软件预测。
差异表达分析时,针对差异表达基因进行的KEGG和GO富集分析,选择哪一个作为参考?
GO和KEGG是两个独立的数据库:
GO数据库的作用是将基因按照它们参与的生物学过程、构建细胞的组分、实现的分子功能等进行分类。
KEGG数据库是将基因按照参与的pathway通路分类。两个均可参考。
后言:
以上是做转录组测序的同学们最常碰到的问题,相信以上解答会为您带来一定的帮助。
更多的信息,可以参见《生工生物有参转录组项目报告》
或者咨询生工生物高通量测序部,
021-57072097、021-57072177
rnaseq@sangon.com
rnaseq2@sangon.com
期待您的咨询!
