
肝癌(肝细胞癌,HCC)是最致命的恶性肿瘤之一,它是世界上第六大最常诊断的癌症和第四大癌症死亡原因。仑伐替尼是一种多靶点酪氨酸激酶选择性抑制剂(TKI),且已被FDA批准作为不可切除的晚期HCC患者的一线治疗药物。但临床试验结果表明,仑伐替尼的临床获益有限,可能是由于耐药性导致的。研究表明,癌细胞可以通过激酶重新布线来获得对各种TKIs的耐药性。因此,探索在使用仑伐替尼治疗后,对重新布线起关键作用的激酶是非常重要的。本文中,作者通过试验揭示了CDK6为仑伐替尼耐药HCC的药物靶点,并强调了化学生物学方法在了解癌症非遗传耐药机制中的应用。
01
仑伐替尼耐药HCC细胞中CDK6上调
作者通过连续给药,获得了PLC/PRF/5和Huh7两种仑伐替尼耐药HCC细胞系。用化学探针标记内源性激酶后,通过SDS-PAGE和荧光扫描比较两种细胞之间的激酶谱差异。结果发现,在仑伐替尼耐药HCC细胞系中观察到一些荧光显著增强的蛋白条带,蛋白条带的分子量在30 - 37 kDa之间(图1d)。接下来,通过质谱分析确定,在增加最高的前5个激酶中,CDK6排名最高。CDK6的分子量为36.9 kDa,与在凝胶内荧光扫描中观察到的蛋白带一致。作者进一步评估了整个蛋白质组的全局磷酸化变化发现,包括CDK6在内的激酶重编程是产生仑伐替尼抗性的关键原因(图1i)。经过临床分析发现,仑伐替尼耐药HCC患者中CDK6较敏感组显著上调,进一步验证了CDK6在HCC中驱动仑伐替尼耐药中的作用(图1j)。
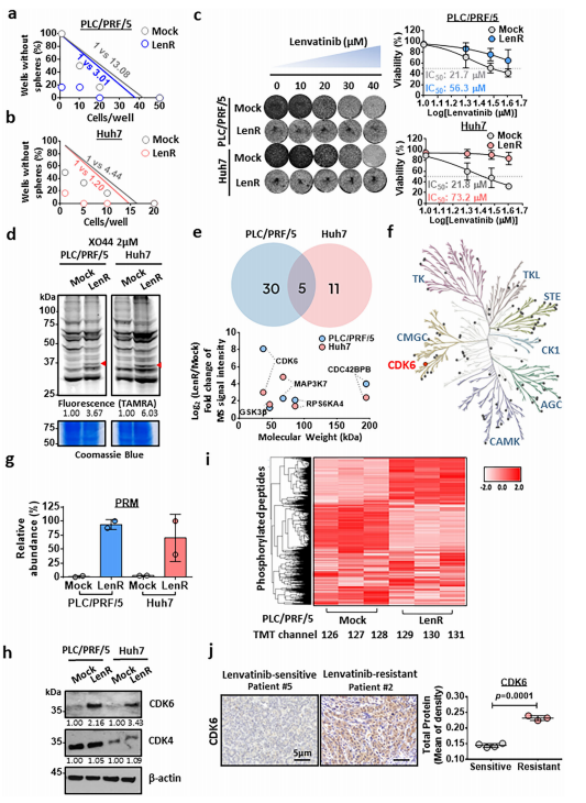
图1. CDK6的激酶谱分析鉴定
02
CDK6调控肝癌细胞干性
CDK6在仑伐替尼耐药HCC细胞中上调(图1),表明其在HCC中调控癌变的作用。为了研究CDK6是否在功能上驱动自我更新和肿瘤形成,作者使用慢病毒敲除和CRISPR基因激活系统来调控HCC细胞中的CDK6表达。在高表达CDK6的HCC细胞系中(包括MHCC97L和Hep3B细胞)进行敲低实验,在PLC/PRF/5细胞进行过表达实验(图2a)。CDK6过表达增强了PLC/PRF/5细胞的干性(图2b)。在体内致瘤性实验中,CDK6的敲低显著降低了肿瘤的大小和形成的数量,PLC/PRF/5细胞中CDK6的过表达显著增强了致瘤性(图2c)。与此一致的是,CDK6的改变改变了四种已知肝脏CSC标志物的表达水平,即CD47、CD90、CD133和EpCAM(图2d)。此外,CDK6被发现在迁移和侵袭中发挥调控作用肝癌细胞的能力(图2e)。
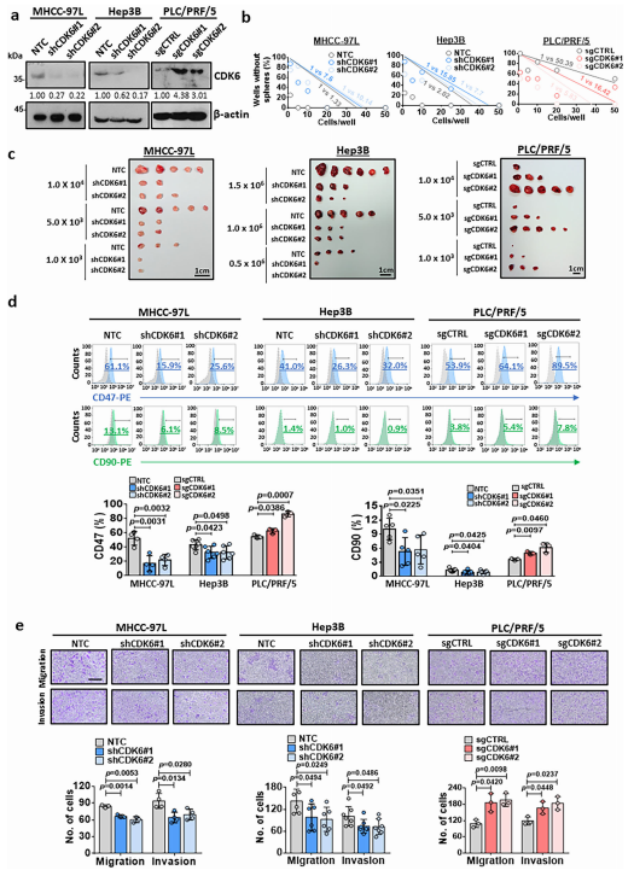
图2. CDK6在肝癌干细胞调控中的关键作用
作者发现CDK6的表达从正常到肝硬化再到进展性HCC阶段逐步增加,这表明CDK6在肝癌发生中的致癌作用(图3a)。作者通过免疫组化评估CDK6在HCC中的表达,发现CDK6高表达患者的无病生存期较短,复发率较高(图3c)。

图3. CDK6在HCC中的临床意义
03
下调CDK6水平可使HCC细胞对仑伐替尼的作用增敏
鉴于CDK6在仑伐替尼耐药HCC细胞中表达上调,作者研究了下调CDK6是否导致HCC细胞对仑伐替尼治疗增敏。MTT试验显示CDK6敲除后,癌细胞生存率降低,对仑伐替尼的耐药性降低。相比之下,在过表达CDK6的细胞中产生耐药性(图4a)。在一组CDK6表达程度不同的HCC细胞中,CDK6的表达水平与仑伐替尼耐药性呈正相关(图4b)。帕博西尼是一种CDK4/6抑制剂,与仑伐替尼联用时显示出协同效应(图4c)。为了进一步证实帕博西尼的增敏作用不依赖于CDK4,作者在仑伐替尼耐药的细胞以及高表达CDK6的细胞中检测了PROTAC(CDK6蛋白降解剂)对CDK6和仑伐替尼的联合作用。结果显示这些细胞中CDK6的降解呈剂量依赖性(图4d)。此外,MTT实验表明,PROTAC与仑伐替尼联合可协同抑制仑伐替尼耐药的PLC/ PRF/5和Huh7细胞以及高表达CDK6的HCC细胞的生长(图4e)。
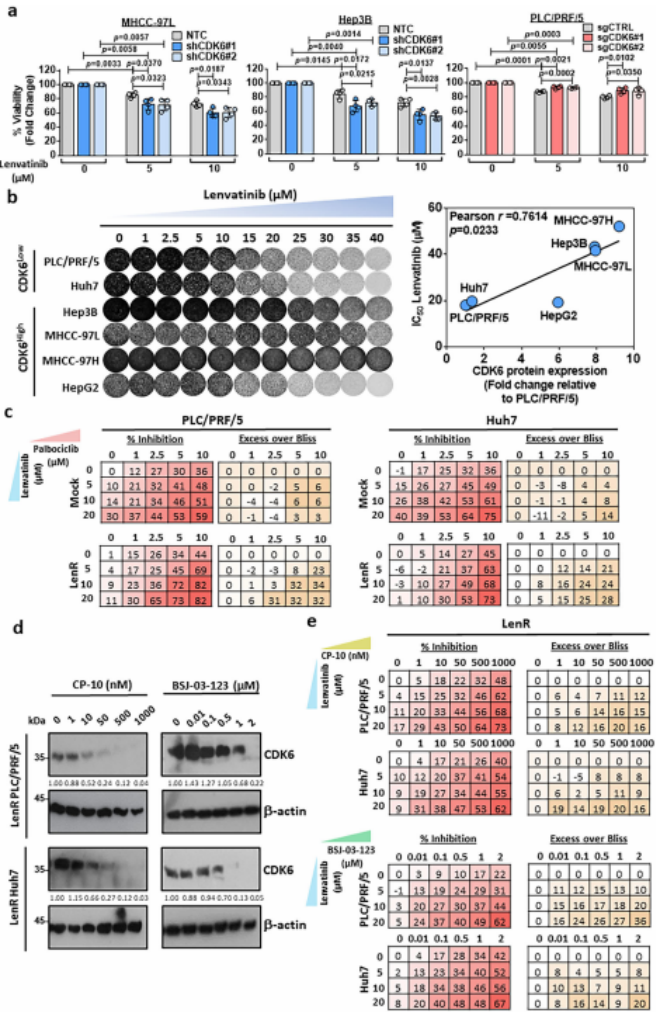
图4. CDK6与仑伐替尼耐药相关
04
ERK/YAP信号通路介导CDK6上调
之前有报道称CDK6表达受YAP1上调,而 YAP 的表达受 ERK1/2调控。在仑伐替尼耐药HCC细胞中,随着CDK6的上调,pERK1/2和YAP的表达也升高(图5a)。为了进一步证实ERK/YAP信号是CDK6表达的上游调节剂,作者用MEK抑制剂处理仑伐替尼耐药的PLC/PRF/5和Huh7细胞后发现,CDK6和YAP的表达下降到与对照组相似的水平(图5b)。在使用YAP抑制剂(CA3)处理的相同HCC细胞中,也观察到类似的CDK6表达水平下降(图5b)。用共聚焦免疫荧光(IF)显微镜进一步证实了这三种蛋白在细胞质和核室中的共定位(图5c)。为了进一步探索ERK1/2如何激活YAP以驱动CDK6的上调,作者使用LATSBiosensor检测了YAP蛋白的上游负调控因子LATS1/2的活性。证实了ERK1/2通过抑制LATS1/2对YAP的调节作用。作者接下来研究了CDK6是否在仑伐替尼耐药HCC细胞中转录上调。
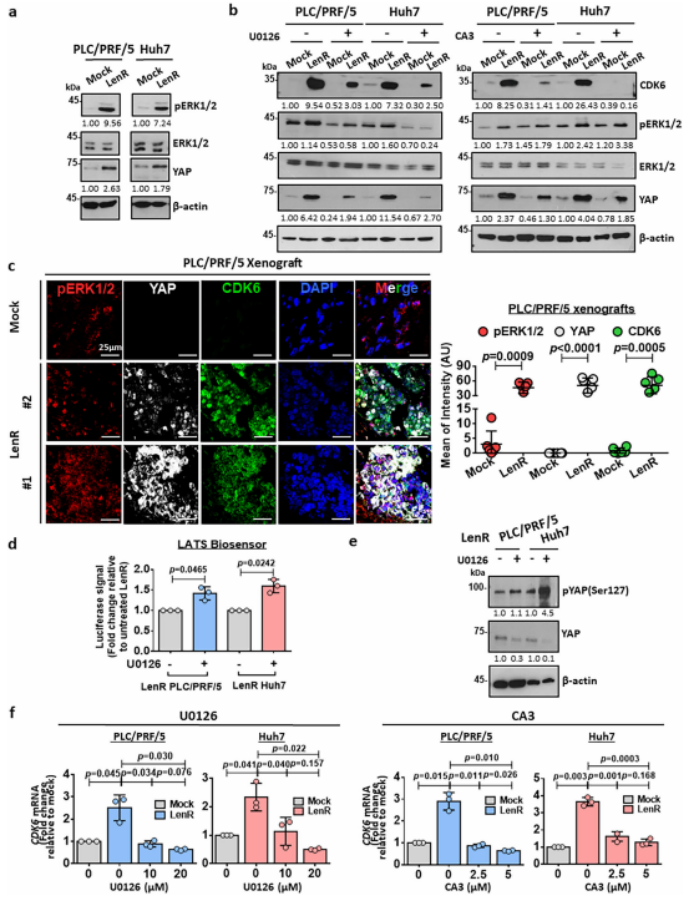
图5. CDK6位于ERK/YAP信号通路的下游
05
CDK6通过结合并磷酸化GSK3β,使Wnt/ β-catenin活化
对仑伐替尼耐药HCC细胞中激酶谱的STRING分析(图1e)显示,CDK6与GSK3β具有潜在的蛋白-蛋白相互作用(图6a)。基于此,作者利用内源性蛋白通过免疫沉淀试验验证了CDK6和GSK3β在这些细胞中存在物理相互作用(图6b)。作者通过体外激酶测定,证实GSK3β是CDK6的直接底物(图6c),并使用共聚焦IF显微镜进一步证实了这两种蛋白的共定位(图6d)。接下来,作者研究了CDK6是否通过直接结合调节GSK3β(Ser9)的磷酸化,结果表明,抑制CDK6可抑制GSK3β (Ser9)的磷酸化和β-catenin的表达,而过表达CDK6后则会出现相反的效果(图6e)。
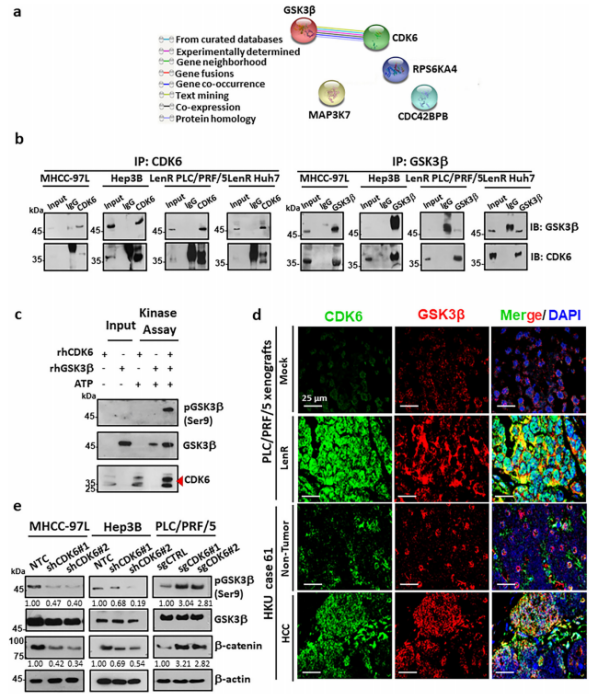
图6. CDK6-GSK3β相互作用的生理意义
鉴于GSK3β在通过泛素化降解β-catenin中的作用,作者发现在HCC细胞中敲除CDK6增加了β-catenin的泛素化(图7a)。减少了 Wnt 靶基因(包括AXIN2、CMYC和CCND2)的转录,减少了 β-catenin 的积累并降低了转录活性(图7b-d)。在过表达CDK6的细胞中,则呈现相反影响(图7a-d)。作者进一步通过抑制仑伐替尼耐药细胞中β-catenin的表达来检测β-catenin是否为CDK6介导的仑伐替尼耐药的下游效应。Annexin-V染色显示β-catenin敲除后细胞存活率降低,仑伐替尼耐药性降低。Cancer Genome Atlas (TCGA)的基因集富集分析(GSEA)显示,在CDK6高表达的HCC患者中,Wnt/β-catenin信号通路显著富集,进一步证实了Wnt/β-catenin是体外CDK6信号通路的主要效应因子(图7e)。以上结果表明CDK6通过直接调节GSK3β的活性调控Wnt/β-catenin信号通路。

图7.CDK6在Wnt/β-catenin信号级联激活中的功能作用
06
帕博西尼联合仑伐替尼在仑伐替尼耐药HCC中抑制效果最佳
作者利用仑伐替尼耐药的细胞的肝癌异种移植物,在体内检测了帕博西尼单用及其联合仑伐替尼的治疗效果。治疗21天后的肿瘤及其相应体积如图8b、c所示。通过观察仑伐替尼未能抑制肿瘤生长,并伴有CDK6高表达。此外,帕博西尼在一定程度上抑制肿瘤生长。帕博西尼联合仑伐替尼发挥协同作用,与对照组相比,对肿瘤生长的抑制最大。作者发现,与第0天的原始肿瘤体积相比,这种联合治疗显著减少了34.9%的肿瘤体积(图8d),且联合治疗组总β-catenin减少(图8e)。此外,与单剂处理和模拟对照相比,联合处理显著抑制细胞增殖,增殖细胞核抗原(PCNA)染色阳性细胞核减少(图8e)。
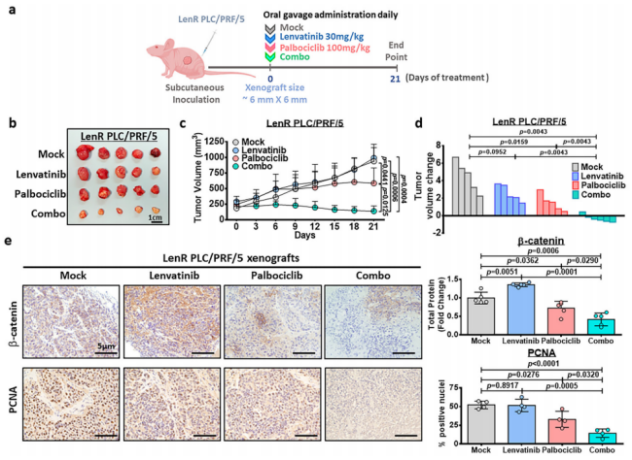
图8.帕博西尼/仑伐替尼治疗对LenR肝癌异种移植肿瘤生长的抑制作用
07
帕博西尼使HCC细胞对仑伐替尼治疗增敏
作者在有免疫功能的小鼠模型中进一步研究了帕博西尼对仑伐替尼耐药HCC细胞的治疗效果。将7-8组的小鼠分为4组,采用相同的处理方法(图9a)。以肝脏重量与体重比及信号强度评价联合用药的疗效,并与单药治疗组进行比较。帕博西尼与仑伐替尼联合治疗对肿瘤的抑制最大。表明帕博西尼治疗与仑伐替尼治疗协同作用,在体内对肝脏肿瘤有效(图9c-e)。与图8e相似,联合治疗组β-catenin和PCNA均下降(图9f)。
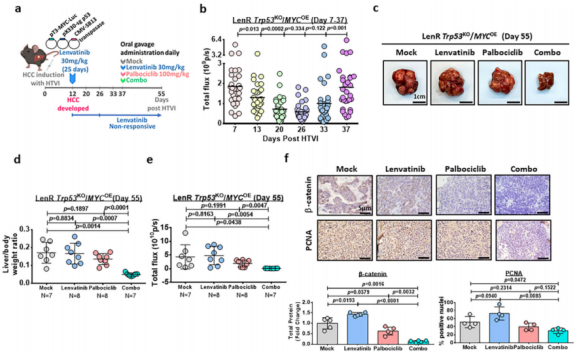
图9.帕博西尼与仑伐替尼联合治疗在小鼠模型中抑制肿瘤生长的作用
作者证明了CDK6在仑伐替尼耐药HCC细胞中β-catenin激活中的作用,并假设帕博西尼将免疫抑制环境转化为免疫炎症环境。通过单细胞RNA (scRNA)测序分析后发现,联合治疗后调节性T细胞(Tregs)减少,记忆效应T细胞亚群增加,而耗竭的CD8+ T细胞亚群减少(图10a, b)。通过CD8α和PD-1标记的耗竭CD8+细胞和CD4和FOXP3标记的Treg细胞的多重荧光免疫组化染色,验证了scRNA测序数据(图10c)。这些发现表明,帕博西尼和仑伐替尼联合治疗后,有利于对HCC肿瘤的免疫抑制。

图10. 帕博西尼与仑伐替尼联合治疗对耐药小鼠模型肿瘤微环境的调节作用
参考文献:
Leung CON, Yang Y, Leung RWH, So KKH, Guo HJ, Lei MML, Muliawan GK, Gao Y, Yu QQ, Yun JP, Ma S, Zhao Q, Lee TKW. Broad-spectrum kinome profiling identifies CDK6 upregulation as a driver of lenvatinib resistance in hepatocellular carcinoma. Nat Commun. 2023 Oct 23;14(1):6699.
来源于优宁维药物研发官网
