
引物设计图解教程 (Guide for Primer Design),实例图解简并引物设计
设计一对合适的引物是PCR扩增目的基因成功的关键,但许多人往往过度依赖PrimerPremier(以下简称 PP)或Oligo等引物设计软件,有时候引物没设计成,却身陷软件之中无法自拔。
其实,不论特异性引物或简并引物,只要掌握了几个关键点,手动也可以设计出一对好引物。如果不是大批量设计引物或设计复杂的引物序列,下面的四个常用工具即可轻松胜任引物设计任务。
本文以马铃薯Y病毒CP基因简并引物设计为示例,分享一些个人经验,希望能起个抛砖引玉作用。专业水平有限,文中有不当之处,敬请各位同行老师批评指正。
相关工具
(1)MEGA5-多重序列比对、选取基因区域、序列编辑;
(2)DNAMAN8-检测两引物的互补性;
(3)0ligo Calc-评估引物的属性;
(4)Web Logo3-直观显示简并碱基。
基本原则
设计一对好的引物,归结起来就是 5'端引物、3'端引物之间以及两者与模板的关系处理得恰到好处。
(1)两引物的序列要与模板的序列紧密互补;
(2)两引物不能在模板的非目的位点发生错配;
(3)两引物之间尽量减少二聚体或发夹结构生成。
延伸原则
(1)引物长度:常用为 18-27bp,最大不可超过 38bp,否则容易导致延伸温度过高不适合 DNA 聚合酶反应 ;
(2)GC含量:一般介于 40%-60%之间,且两个引物之间的 GC 含量相差不能过于悬殊;
(3)碱基分布:随机分布最佳,但避免连续的 GC,GC 富集区容易导致错误引发反应。
特别注意
5'端引物的作用主要限定 PCR 产物的长度,对扩增特异性影响不大;引物的延伸是从 3端开始的,所以3"端引物是影响特异性扩增的最关键因素,因此,在实际设计过程中,设计3'端引物时,需要综合考虑以下几个内容。
(1)不要终止于密码子的第3位;
(2)末位碱基避免使用碱基A;
(3)避免出现3个以上连续的G或C,如GCG或CCC或GGG;
(4)AG的绝对值不可超过9;
(5)与非特异扩增的序列同源性不能超过 70%或有连续8个互补碱基同源;
(6)不能进行任何修饰。
设计流程
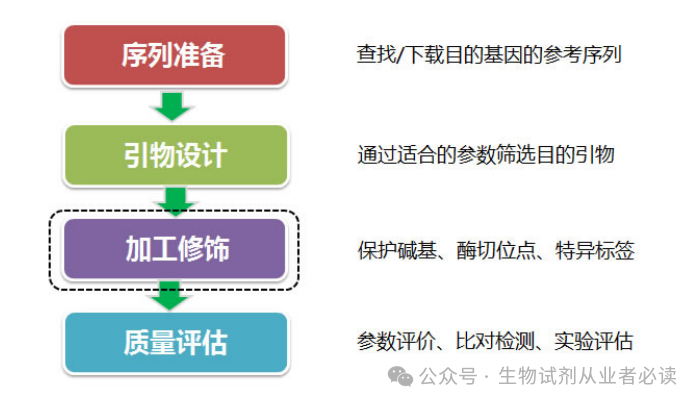
示例背景
马铃薯 Y病毒( Poialo vinus Y,PVY)是侵染马铃薯、烟草、辣椒等茄科作物并造成严重危害的病毒之一,广泛分布全球各马铃薯种植区。RT-PCR 技术具有高度的特异性和灵敏性等特点,已经成为 PVY 检测最常见的方法。但由于PVY 株系分化严重,不断有新的重组株系产生,单一的特异性引物无法适应 PVY 不同株系的检测需求,需要设计一对简并引物以能够满足生产上的检测需求。
详细图解
1、序列准备:
(1)GenBank 下载 PVY 全基因组序列
(2)由于基因序列比较大,且数量多,推荐用 MAFFT 多重序列比对(3)扩增片段区域选择,CP 基因长度及位置如下图所示:

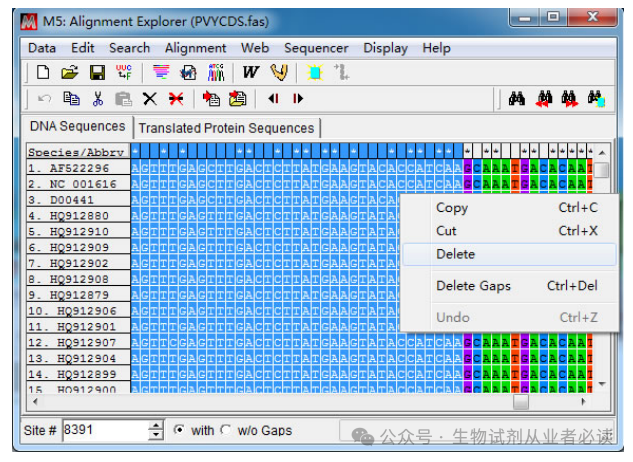
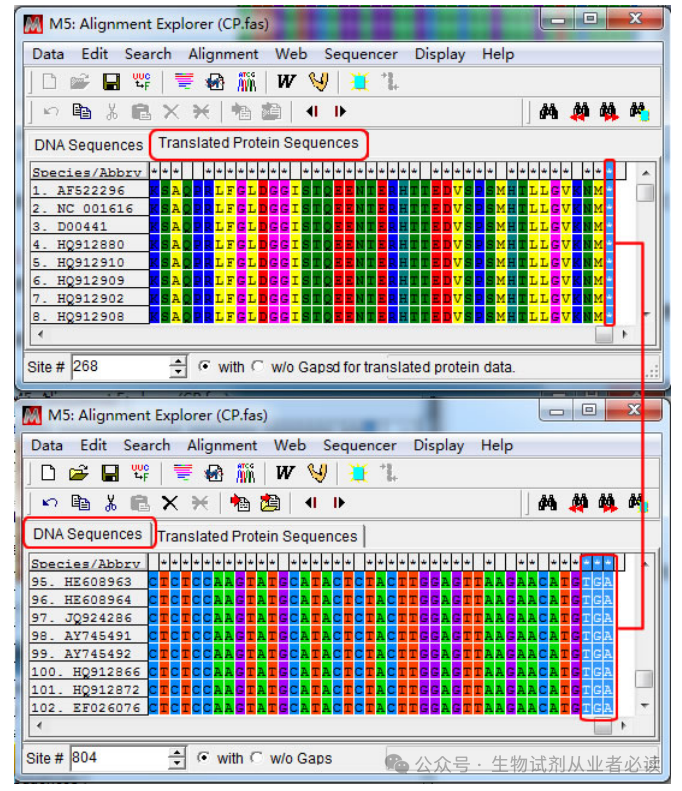
先将光标定位在第一条序列任意位置,然后在左下角"Site"处直接输入 CP 基因上游分界点位置(8391)后回车。接着点击“Speicaes/Abbrv”和 8391那一列的交界点时按下键盘上“Shi”不放,移动光标到“1”那一列,此时点击鼠标右键“Delete”删除冗余序列。
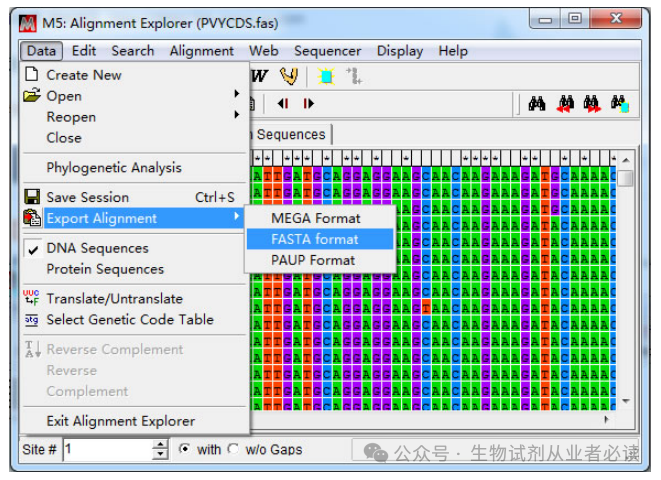
裁切后得到 CP 基因编码区序列“Export Alignment”导出 CP基因序列 推荐fasta格式)
2、引物设计
推荐先设计 3端引物,至于原因,上面的【特别注意】中已提到,这里不再赘述,
(1)选择引物序列
综合考虑引物设计原则及特别注意,在 CP 基因 3'端位置选取一段合适的序列(784-803bp),804bp 位置为A且是第三个密码子位置,所以弃去未位的 A碱基。
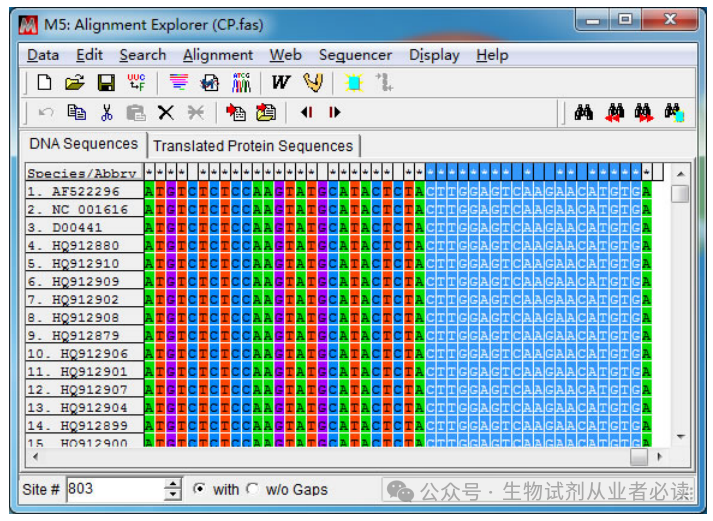
是否位于密码子的第3位,可以通过“Tranlated Protein Sequences”-“DNA sequences切换查看,如下图,CP 基因的末端3个碱基是终止密码子所在的位置,A 是第三位密码子
(2)生成 SeqLogo
选定序列后,参考上述步骤,删除冗余序列,保留 CP 基因 3'端序列,同样导出为 Fasta文件,将序列粘贴入 Web Logo3(http://weblogo.threeplusone.com/create.cgi)的文本框内或直接上传序列文件,设置相关参数后点击“Create”生成 Seq Logo 格式的文件 CP-3.fas。
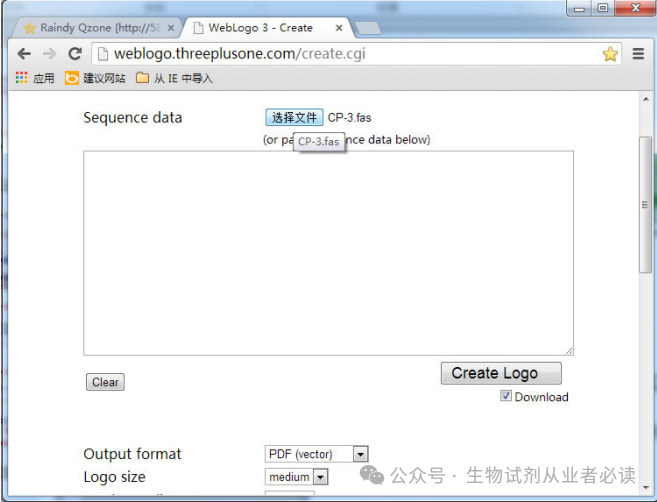
参数设置:“Output fommat’( 输出格式)推荐用“PDF”,“Color scheme”(颜色方案 )推荐用“Classic (NA)”,设置完毕,点击右下角“Create Logo“即可得下图的 SeqLogo 格式文件。
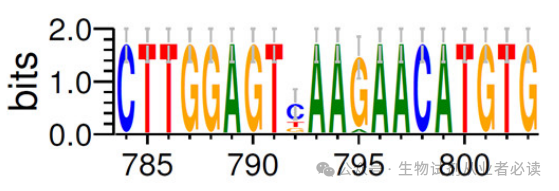
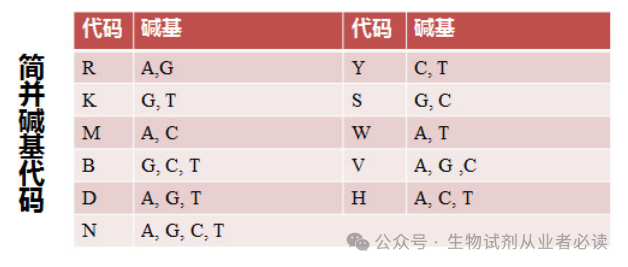
通过 Web Logo 生成的多重比对图片可以很直观得到 3'端序列为CTTGGAGT(C/T/G)AA(G/A)AACATGTG,查询简并碱基代码表(下图),可知 C/T/G=BG/A=R,简并度=3 x2=6,整理后3端序列为 CTTGGAGTBAARAACATGTG,故而需要合成的3端引物序列:CACATGTTYTTYACTCCAAG (与原序列是反向互补关系)。
3、质量评估
(1)参数评估
将初步得到的引物序列,粘贴入 Oligo Calc 的文本框内,按下“Calculate”按钮,得到引物的相关参数,如:长度、GC 含量、Tm 值等信息。
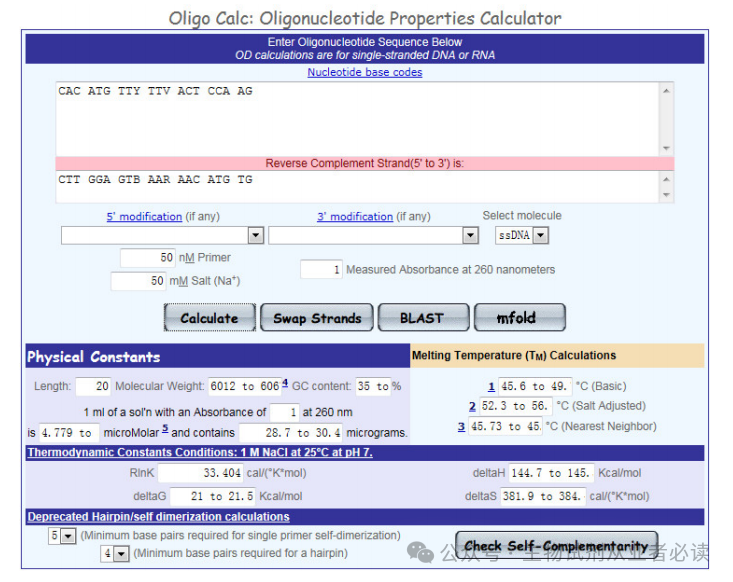
(2)自身互补性(Check Self-Complementarity点击“Check Self-Complementarity”,检测引物自身互补性,主要查看“Potential hairpinformation”、 “3’ Complementarity”、 “All potential self-annealing sites are marked in red (allowing1 mis-match)”下方显示内容,如果三项均为“none“,则说明引物自身不会形成发夹结构且引物自身不会互补,引物没问题。、
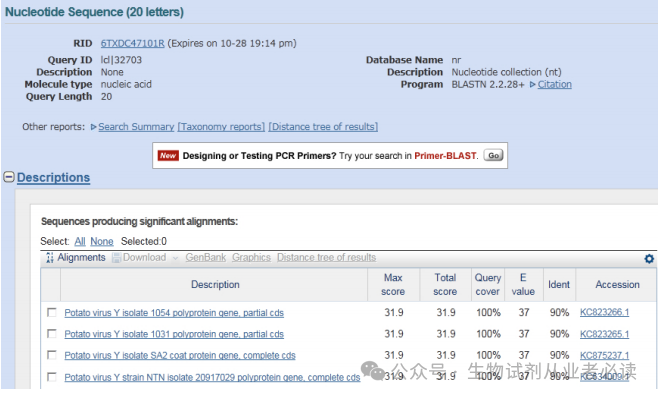
(3)BLAST 比对回检
点击“Blast”可以对引物进行 BLAST 比对检测,结果显示,都是 PVY CP 基因相关的序列,表明所设计的引物特异性良好。
(4)两引物互补性检查
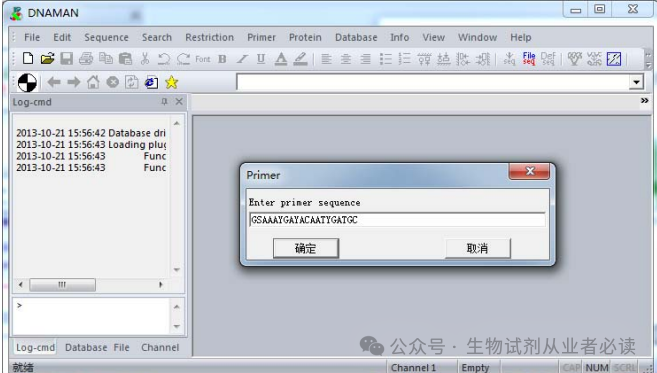
同样方式,得到 5'端序列:GSAAAYGAYACAATYGATGC,根据引物设计原则,需要检测两引物之间是否存在序列互补。
打开 DNAMAN ,依次在“'Primer”“Load primer”,粘贴任意一端引物序列,如:5'端序列-GSAAAYGAYACAATYGATGC,确定。
接着,在“Primer”.“Two primers Complementarity”.“Second Primer From Input”,粘贴入另一端序列,这里为 3'端引物序列:CACATGTTYTTVACTCCAAG,如下图。
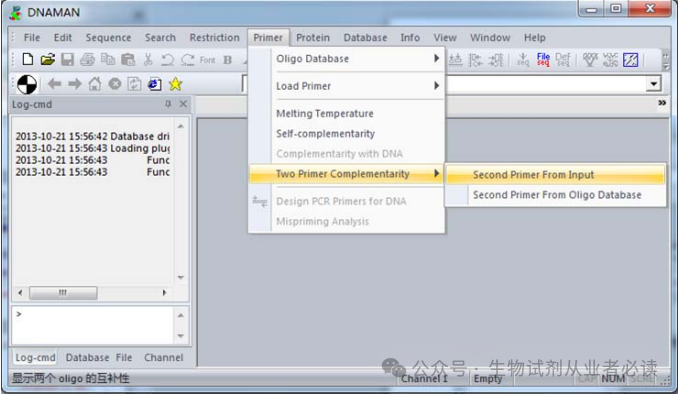
结果显示,两引物间仅有3个碱基互补,且自由能仅为1.8 Kcal/mol。

(5)实验验证
PCR 扩增采用 50uL 反应体系为:10 xTransTag HiFi Buffer Ⅱl 5 μL ,2.5nM dNTPS4 μL5'端引物(10 μmol/L)2μL,3端引物(10 μmol/)2 μL ,ddH20 34.75 μL ,TransTag HiFiPolymerase ( 5 U/μL )0.25 μL ,CDNA 2 μL。
PCR扩增程序条件为:94℃预变性 5min ,94℃变性 30s,55℃复性 30s,72℃延伸 1min共 30 个循环,最后一轮循环后 72℃延伸 10min。反应结束后,1%琼脂糖凝胶电泳检测 PCR扩增产物,如下图所示。
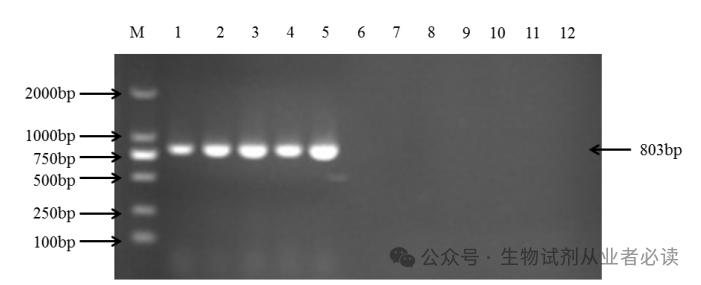
M.DNA分子量标准(100bp);泳道1-5:PVY的不同株系(C、O、N、NTN、N-Wi);泳道6-7:阴性对照(健康马铃薯)和空白对照;泳道8-12:PVX、PVM、PVA、PVS、PLRV
