
生物药品的获批需要在分子水平上进行深入的表征,包括氨基酸序列的确认以及对翻译后修饰(PTMs)的综合评估。糖基化是其中一个关键的质量属性,因为它可能影响药物的疗效和安全性。关于糖基化的信息可以通过传统的肽图的方法获得,但通常仅限于聚糖或糖肽水平。
目前的分析方法包括释放的聚糖或糖肽的表征,提供平均分子组成的详细信息。另一种方法是,通过测定完整的蛋白来揭示特定蛋白形式的存在和相对丰度。在变性条件下的质量测定仅限于结构相对简单的样品,这是因为该状态下会观察到的信号簇较多重叠。相比之下,在天然条件下,蛋白质折叠被保留,信号可以被分离,从而反卷积这些宽信号集群,以表征非共价蛋白复合物。作者在本文中证明了在天然条件下质谱解析依那西普高度复杂的糖基化模式的适用性。
01
依那西普完整水平的天然质谱分析。
作者首先测量了完整水平依那西普的质量,得到完整蛋白质变体的质量范围为125至131 kDa。考虑到依那西普氨基酸序列的理论平均质量为102,160 Da,剩余质量为23,000~ 29,000 Da,对应蛋白质的聚糖部分。
随后,作者利用糖苷酶和蛋白酶,依次降低样品的复杂性和分子大小,以获得依那西普在不同结构层次上确定的所有修饰组合。作者设计了几种方案:(i)用唾液酸酶去唾液酸化,(ii)用PNGase F去N糖基化,(iii)两者结合,(iv)用唾液酸酶和O-糖苷酶双酶切,(v)用IdeS酶切将依那西普分解成依那西普和Fc结构域,(vi)用胰蛋白酶或AspN酶切成糖肽,然后对所得产物进行质谱分析。
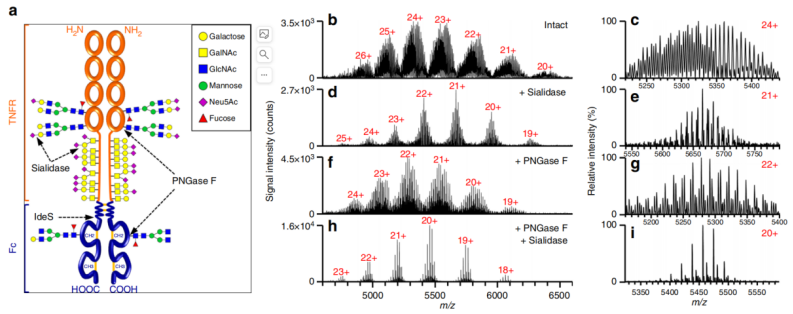
图1
02
去除N -聚糖后O-聚糖的测定。
质谱分析显示,糖苷酶消化后,信号向较低的m/z方向移动,并降低了复杂性(图1d - i)。去除N -聚糖和唾液酸残基所得到的糖型只是核心O-聚糖的数量不同(图1h)。因此,双糖苷酶消化的反卷积光谱中最丰富的峰的质量相差365.1 Da,这是由于一个己糖(Hex)和一个N -乙酰己糖胺(HexNAc)残基,具有核心1 型O-聚糖结构的特征(图2a)。
与PNGase F/唾液酸酶双消化相比,PNGase F处理依那西普的光谱显示出更高的复杂性,这是由于O-聚糖的唾液酸变体(图1f, g),对于给定数量的附着核心O-聚糖,可以看到一系列分子质量相差291.3 Da的信号(图2b)。考虑到在双糖苷酶酶切中观察到的O聚糖的相对丰度(图2a),相应的O-聚糖的唾液化变体可以被注释(图2b)。
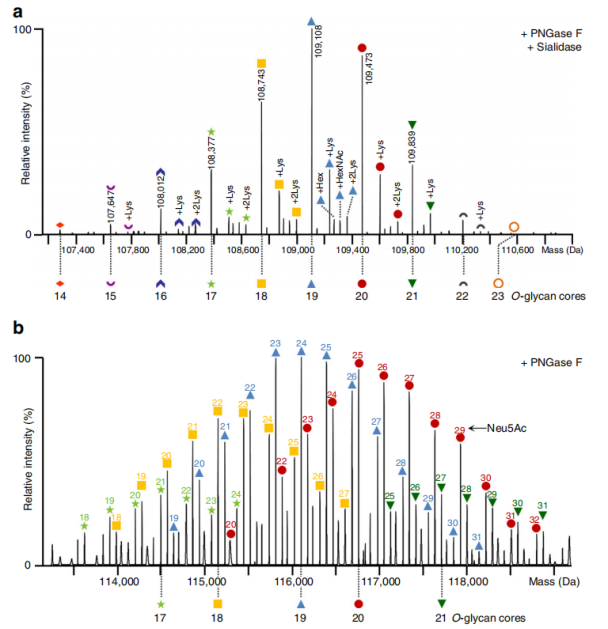
图2
03
依那西普结构域N -糖基化的评估。
作者使用IdeS蛋白酶对依那西普进行酶解,在铰链区域酶切蛋白质,随后通过亲和纯化分离依那西普和Fc结构域。为了获得依那西普 N -聚糖变体的信息,作者首先使用O-糖苷酶去除O-聚糖,所得到的峰仅包含缺乏唾液酸的N -聚糖变体(图3a)。
为了精确定位依那西普结构域上的N -聚糖结构,作者用AspN消化后获得的糖肽生成了一个特定位点的N -聚糖文库。光谱中存在的大多数峰可以明确地归因于特定的N -聚糖组合,即基于从糖肽数据中获得的分数丰度的N -糖型。除了含有两个残余O-聚糖的物种外,还可以检测到不含或多达三个核心O-聚糖的变体(图3a)。
04
Fc结构域N -糖基化的测定。
通过亲和纯化天然质谱,作者成功地获得了二聚体Fc结构域的分子质量数据(图3b)。在相应的色氨酸糖肽丰度分析中,最显著的糖型为A2G0F/A2G0F(50,464.7 Da)、A2G0F/ A2G1F(50,626.7 Da)以及等压变体A2G1F/A2G1F或A2G0F/A2G2F(50,788.7 Da)(图3b)。其中,N-糖型A2G1F/A2G1F等压变体的丰度估计为70%,而A2G0F/A2G2F的丰度则为30%。此外,我们还观察到c端赖氨酸变异的相对丰度分别为~ 24.3%(+赖氨酸)和2.8%(+2赖氨酸)。这些数据与整体蛋白的c端色氨酸SLSLSPG(88.1%)和SLSLSPGK(11.9%)的数据相一致。
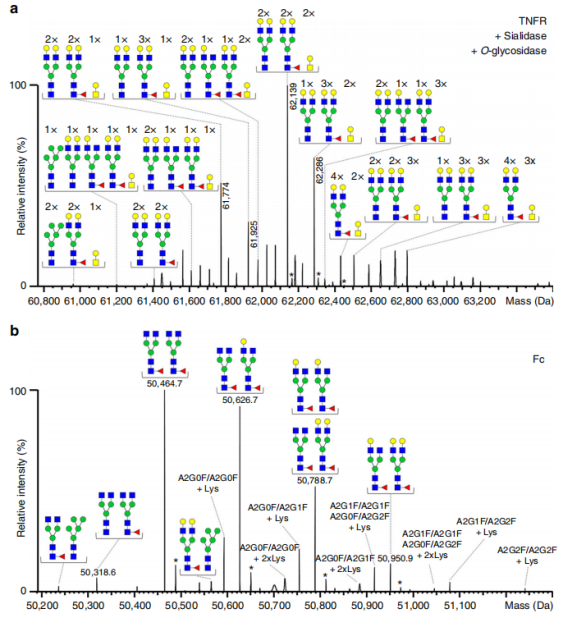
图3
05
整合全蛋白水平的糖基化数据。
作者在亚基水平上鉴定了N-聚糖组合,如图3a和b所示。这些N-糖型在唾液苷酶/O-糖苷酶处理后的依那西普中得到了注释,如图4a所示。值得注意的是,经过酶解处理后,最丰富的N-糖型(112,732 Da)是由最丰富的依那西普N-糖型(62,139 Da,图3a)和第二丰富的Fc糖型(50,627 Da,图3b)组合而成的。在蛋白质水解过程中,这两个糖型结合了两个水分子。同样地,其他糖型在唾液酸酶/O-糖苷酶处理依那西普后,也可以通过整合依那西普和Fc结构域的N-聚糖数据来识别。将图3a和3b所示的数据进行合并,可以识别出六种不同的N-聚糖组合。这些N-聚糖组合可以被多达三个剩余的核心O-聚糖所取代,如图4a所示。此外,注释与基于位点特异性N-糖肽文库的糖型分配一致,验证了在不同消化水平下数据整合的一致性。
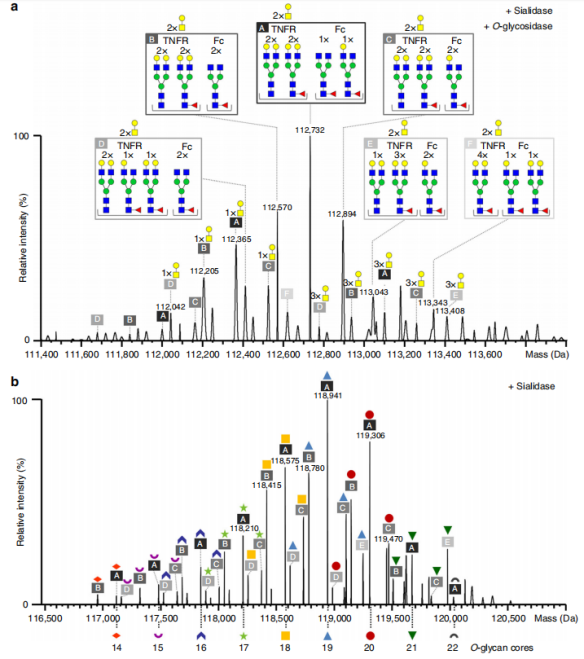
图4
就完整的(未经处理的)依那西普的糖基化而言,图1b和1d所描述的光谱的比较清楚地表明,加入唾液酸后复杂性增加。在整个蛋白质水平上对特定糖型的分配通常是模糊的,因为对多种糖变体的分辨率不足,它们的质量差异很小。然而,完整的依那西普的光谱显示出高度独特的峰型,这促使人们对开发的用于评估不同依那西普生产批次中聚糖一致性的天然质谱方法进行评估。
06
用天然质谱对完整依那西普进行的分析。
如图5所示,揭示了92到122个不同的信号,提供了产品异质性的详细和特征指纹。此前曾报道过依那西普生产批次的质量变化,其原因是与更改前批次相比,更改后批次的A2G2F N -聚糖结构减少以及c端赖氨酸变体增加。作者还观察到变化前后材料的完整质量差异(图5a),而两个变化后批次的光谱具有高度可比性(图5b)。观察到的差异可能主要归因于N -聚糖水平,因为通过PNGase F去除N -聚糖后,依那西普批次的天然光谱更具可比性,与先前发表的N -糖基化差异一致。
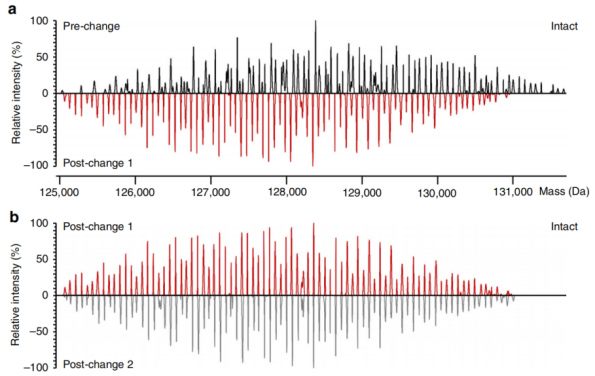
图5
这篇文献的结果展示了天然质谱与酶解技术相结合的方法在研究生物药物糖基化形式异质性方面的应用价值,并为推动生物药物的研究和发展提供了重要的理论基础和技术手段。
来源于优宁维药物研发官网
