多细胞生物严密地协调自噬和细胞死亡,以维持发育、分化和许多细胞类型的稳态[1]。基于形态学、生化和分子机制差异,调节性细胞死亡主要分为凋亡形式和非凋亡形式[2]。铁死亡是一种非凋亡性细胞死亡,主要依赖于铁介导的Fenton反应和随后细胞内过量的脂质过氧化[3]。铁死亡损伤涉及多种病理情况,特别是癌症和组织损伤。GPX4(谷胱甘肽过氧化物酶4)是system xc-的下游抗氧化酶,参与细胞膜中磷脂氢过氧化物的清除,从而对铁死亡具有细胞保护作用。抑制GPX4活性或下调GPX4表达可诱导铁死亡。虽然GPX4的抗铁死亡功能已被广泛研究,但GPX4的调控机制仍不甚清楚。
铜是人体生理必需的微量元素,其代谢的改变与多种疾病发病密切相关。在大量不同类型的癌症患者中,包括淋巴瘤、乳腺癌和肺癌,都观察到血清和肿瘤铜水平升高。并且,铜螯合对肿瘤发展和血管生成具有潜在的抑制作用[4]。然而,过量摄取铜也会导致细胞损伤和死亡。特别是,细胞内铜的超生理水平通过诱导活性氧(ROS)或抑制蛋白酶体功能来触发凋亡[5]。此外,细胞内铜的积累可能导致一种特定的细胞死亡方式,称为“铜死亡”,其不同于凋亡、坏死和铁死亡[6]。在以往的研究报道中,铜似乎可以通过几种依赖于环境而不同的死亡途径来杀死细胞,但铜是否调节铁死亡尚未明确。
该研究报道了铜在体外和体内促进铁死亡敏感性的新作用。首次证明了铜直接与GPX4蛋白结合,导致GPX4聚集体的形成和随后的GPX4自噬降解。另外还发现TAX1BP1(Tax1结合蛋白1)是GPX4蛋白降解的自噬受体。这些发现为自噬依赖性铁死亡的机制提供了新的见解。
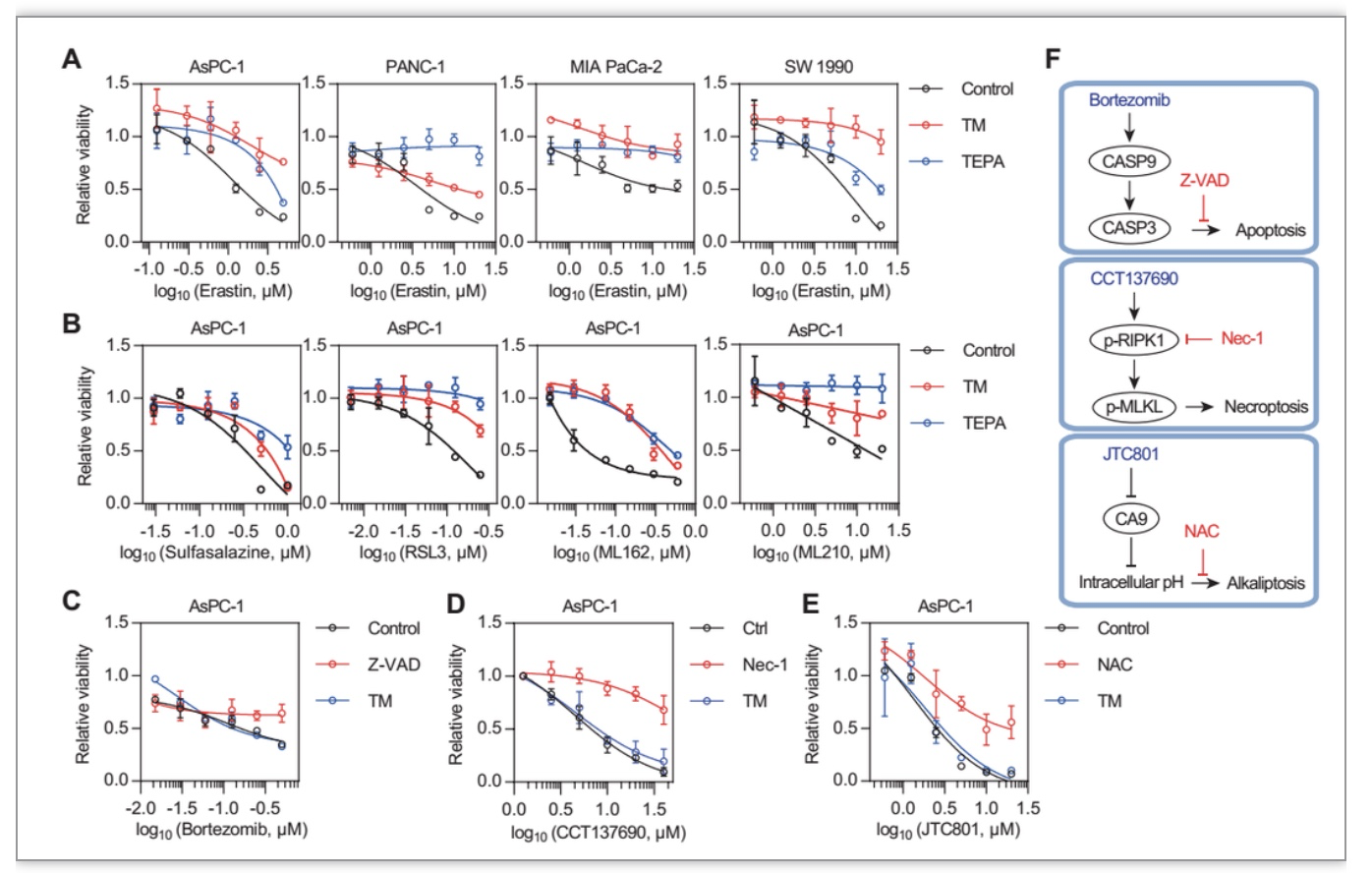
为了确定铜是否调节铁死亡,作者使用铜螯合剂TM和TEPA处理四种胰腺癌细胞系Aspc-1、PANC-1、MIA PaCa-2和SW1990细胞,确定铜螯合剂TM和TEPA能够抑制铁死亡诱导剂erastin引起的细胞毒性(图1A)。并且,TM和TEPA对多种铁死亡诱导剂包括RSL3、erastin、ML162、ML210、SAS等诱导的细胞死亡均有抑制作用(图1B)。TM和TEPA只能抑制铁死亡而不抑制凋亡、坏死和碱死亡(图1C-F)。同时,作者发现TM和TEPA可以抑制erastin诱导的脂质过氧化,但对细胞内铁离子没有影响。与经典的铁死亡抑制剂liproxstatin-1和DFO不同,TM和TEPA在体外不具有抗氧化性,在体外也不能够结合铁离子。以上结果证明TM和TEPA可以抑制铁死亡。
(图源:Qian Xue, et al., Autophagy, 2023)
作者在细胞培养基中添加铜离子之后,发现Cu2+会对低剂量的erastin处理造成细胞死亡(图2A)和脂质过氧化(图2B),这一过程被铁死亡抑制剂Fer-1阻断(图2C)。当使用铁螯合剂DFO去除游离Fe2+时,向erastin中加入Cu2+并不能诱导细胞死亡(图2E),说明Cu2+不能取代Fe2+介导Fenton反应(图2F)。作者进一步研究了Cu2+对GPX4蛋白表达的影响,结果显示,TM阻止了Erastin诱导的GPX4蛋白下调,而Cu2+则促进了GPX4蛋白下调(图2F,G)。同时,GPX4 mRNA水平不受影响(图2H,I)。重要的是,Cu2+联合erastin诱导了AsPC-1细胞GPX4蛋白的泛素化(图2J),说明铜可能促进了GPX4蛋白的降解。既往研究表明,GPX4蛋白降解可通过蛋白酶体和自噬途径介导。随后作者发现只有自噬的抑制剂CQ可以阻止GPX4蛋白降解,而不是蛋白酶体BTZ(图2K)。铜还可以促进GPX4蛋白的多聚化(图2L)。因此,自噬可能参与了GPX4蛋白的降解。
(图源:Qian Xue, et al., Autophagy, 2023)
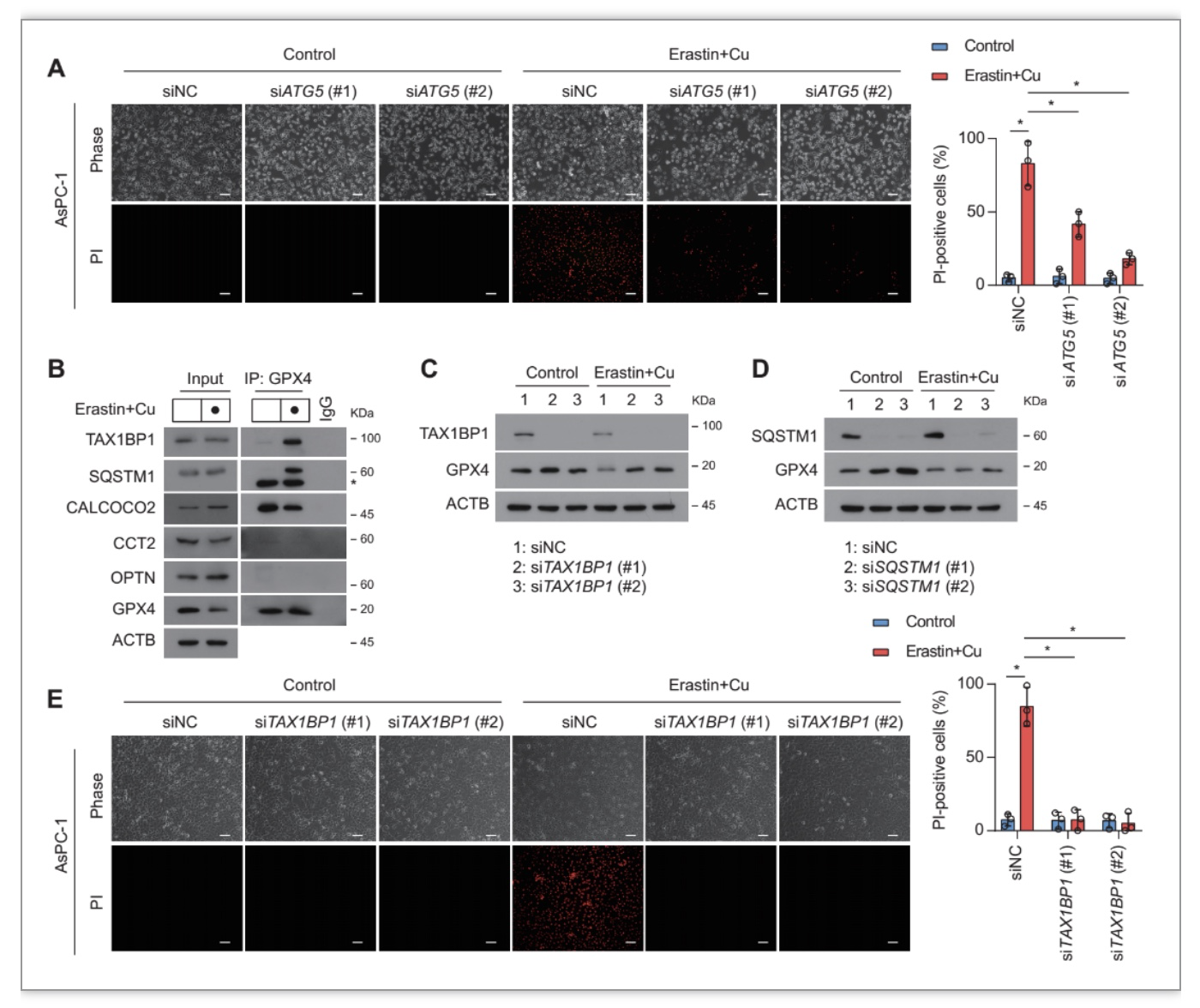
作者推测自噬分子参与GPX4蛋白降解,作者发现铜和erastin的联合作用显著激活了自噬。此外,ATG5的下调也减弱了Cu2+联合erastin诱导的铁死亡(图3A)。紧接着作者发现Cu2+联合erastin选择性地增加了GPX4与TAX1BP1和SQSTM1的结合,但没有增加与其他自噬受体CALCOCO2、CCT2或OPTN的结合(图3B)。同时敲低TAX1BP1(而非SQSTM1/p62)抑制GPX4蛋白降解,并且降低了细胞对铁死亡的敏感性(图3C-E)。以上结果证明TAX1BP1是GPX4蛋白降解的关键自噬受体。
(图源:Qian Xue, et al., Autophagy, 2023)
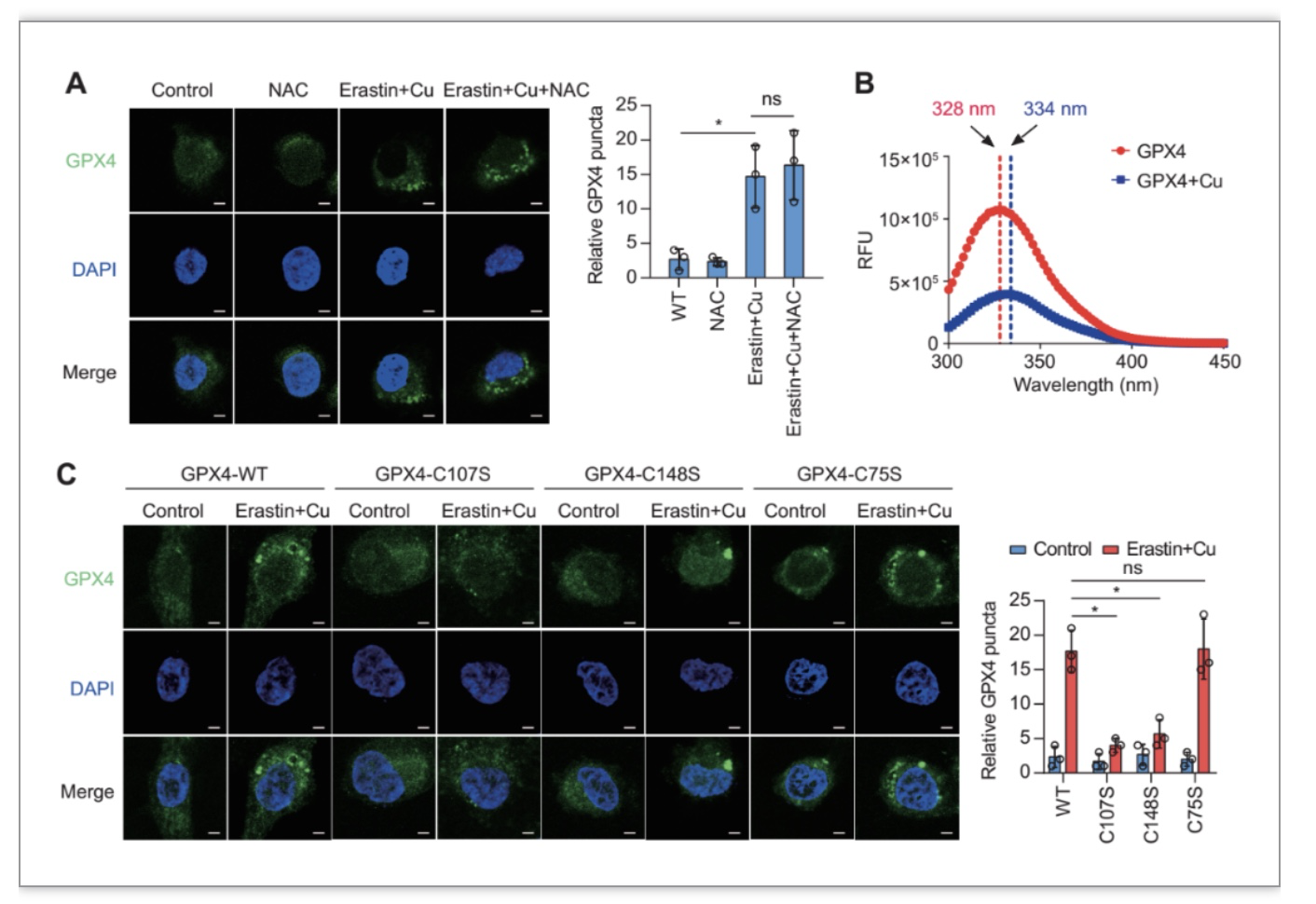
由于重金属诱导的ROS生成是细胞杀伤的重要机制,为了排除Cu2+介导的ROS生成参与这一现象的可能性,作者使用ROS清除剂NAC发现未能抑制Cu2+联合erastin诱导的GPX4聚集(图4A)。接下来,作者研究了Cu2+是否直接与GPX4结合以诱导GPX4聚集。为了验证这一假设,随后作者进行了基于重组GPX4蛋白荧光光谱的变化的研究,发现在Cu2+处理下GPX4发生了明显的光谱移位(从328 nm到334 nm),这表明铜与GPX4之间可能存在直接的相互作用(图4B)。先前有研究发现Cu2+可与半胱氨酸残基相互作用,而GPX4中有两个表面可接近的半胱氨酸残基C107和C148,理论上可能与Cu2+结合。用丝氨酸残基取代这些半胱氨酸残基的位点定向突变表明,C107和C148都是Cu2+诱导的GPX4聚集在Cu2+联合erastin作用下发生的必要条件(图4C)。相比之下,未表面暴露的C75突变未能影响Cu2+诱导的GPX4蛋白聚集(图4C)。这些结果表明Cu2+选择性地与GPX4的半胱氨酸残基C107和C148结合。
图4 铜诱导的GPX4聚集依赖于GPX4的半胱氨酸残基
(图源:Qian Xue, et al., Autophagy, 2023)
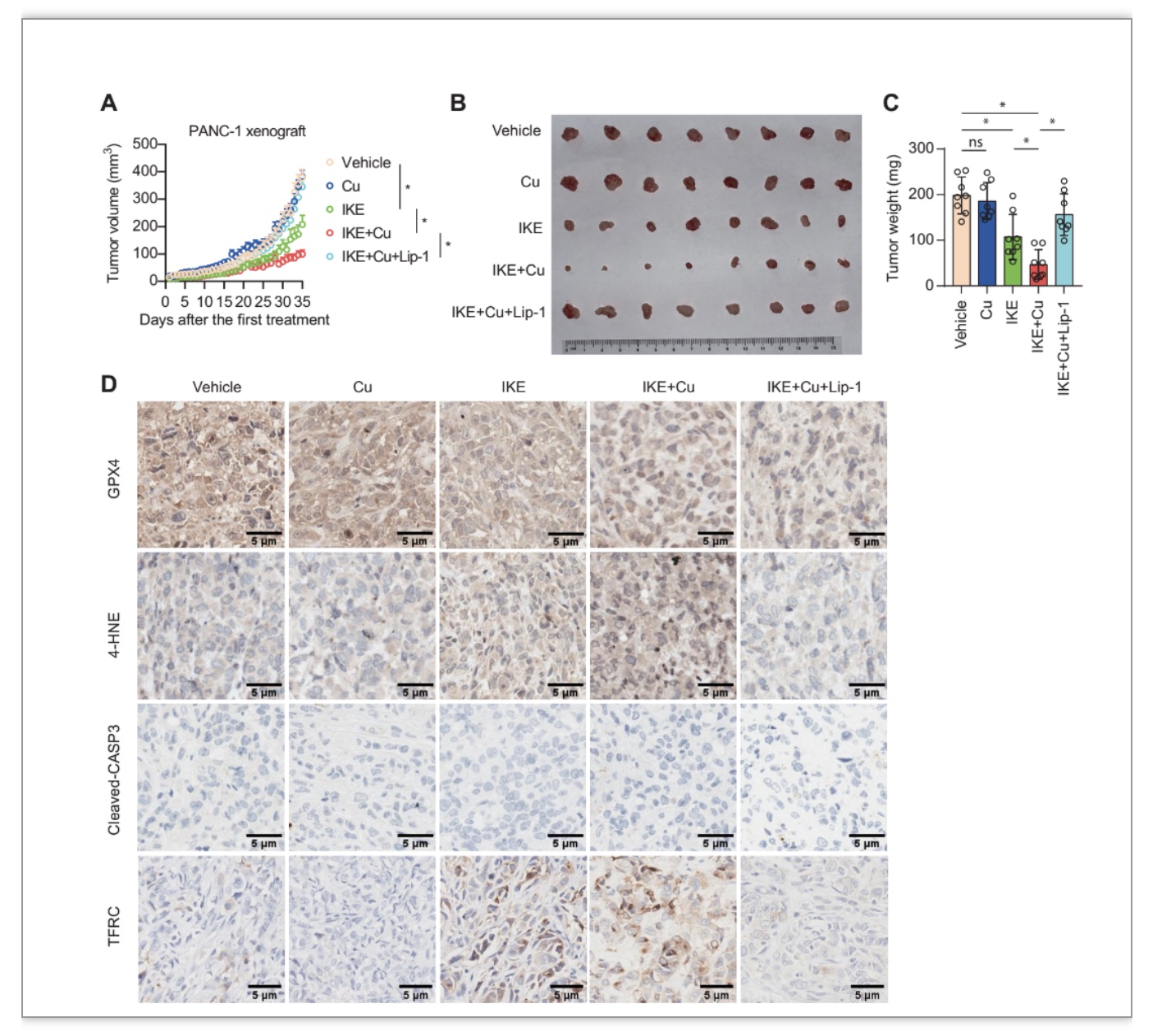
作者进一步探究了铜对裸鼠在体移植瘤中铁死亡的影响。与细胞实验中的发现一致,相比对照组,PANC-1异种移植到IKE治疗,铜增强了IKE治疗效果,这种效果被铁死亡抑制剂Lip-1逆转(图5A-C)。免疫组化染色结果显示,铜离子和IKE联合使用对GPX4水平的抑制强于单一药物治疗(图5D)。此外,联合治疗增加了肿瘤中4-HNE的水平,但没有增加Cleaved caspase 3的表达水平(图5D)。然而,铜并不能影响erastin诱导的TFRC和Fe2+水平(图5D),说明铜并不影响肿瘤中与铁死亡相关的铁代谢。作者还评估了铜在急性胰腺炎中的作用,通过实验证明:铜有助于铁死亡诱导的急性胰腺炎。综上,这些结果证实了铜可以在在体内促进铁死亡的发生。
(图源:Qian Xue, et al., Autophagy, 2023)
(图源:Qian Xue, et al., Autophagy, 2023)
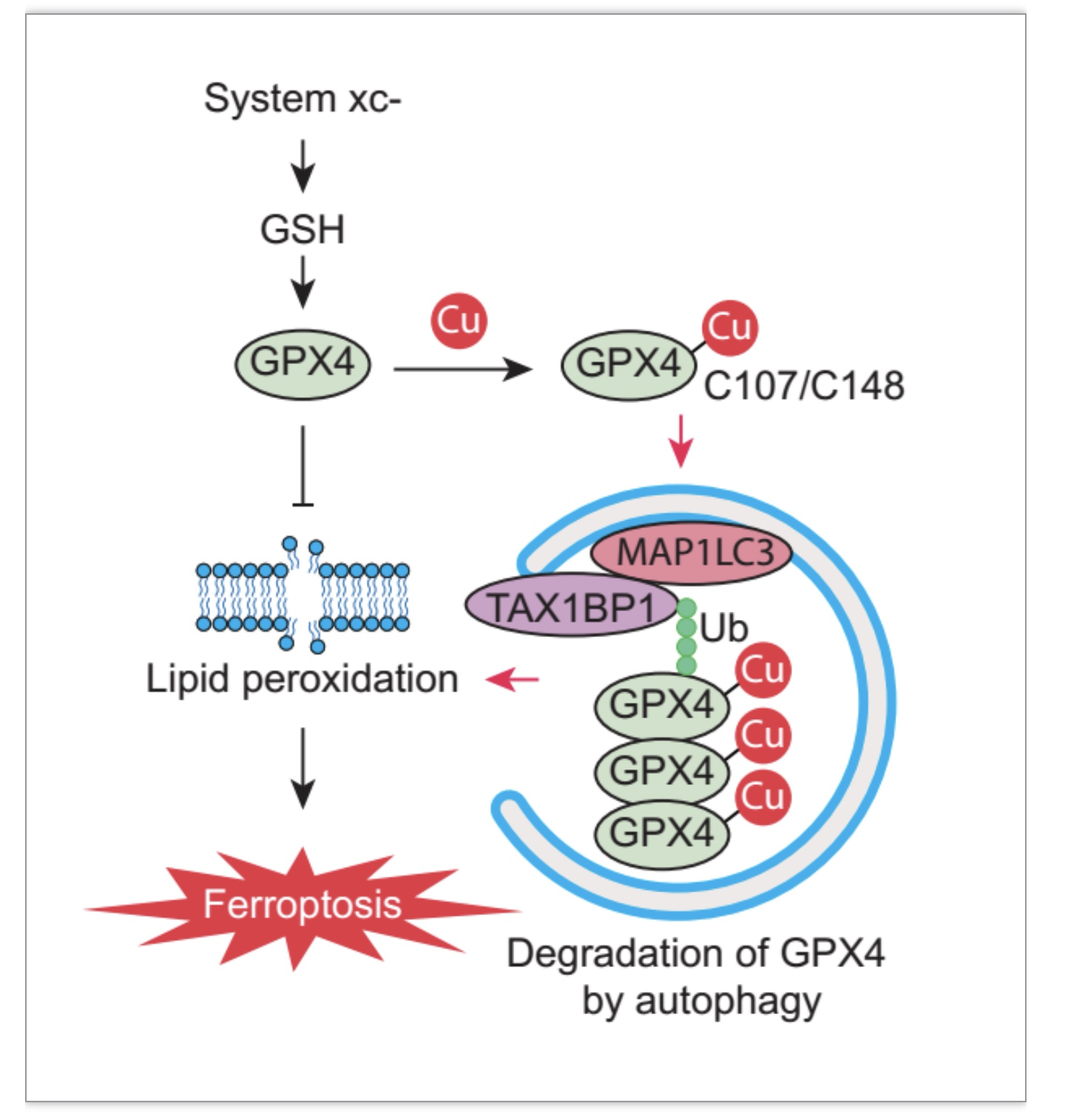
综上所述,该研究揭示了铜离子的一种新的促铁死亡作用及机制,即铜离子与GPX4蛋白特定半胱氨酸残基的结合并促进依赖自噬受体TAXIBP1的GPX4自噬降解(图6)。作者的发现可能为通过给予额外的铜离子来增强癌细胞的铁死亡,以及通过铜离子螯合来抑制组织损伤性铁死亡提供潜在的治疗策略。