

该研究首先评估了BT-Br对DU145细胞中CAT的影响。结果表明,将不同浓度的BT-Br与DU145细胞共同孵育48h后,胞内ROS的浓度显著提升,且CAT mRNA的相对水平和蛋白含量均明显降低,表明BT-Br可以有效抑制DU145细胞中CAT的活性,并最终影响CAT的表达。接下来,为了更好地了解BT-Br与CAT的结合模式和结合位点,该研究通过质粒构建和蛋白表达纯化得到高纯度的人CAT,将其与BT-Br共结晶后,获得分辨率为2.2 Å的CAT-BT-Br共晶体(PDB ID:8HID)。晶体解析结果显示,BT-Br位于CAT的NADPH结合位点(图1左)。这是第一个结合在该位点,并表现出高抑制活性的小分子CAT抑制剂。
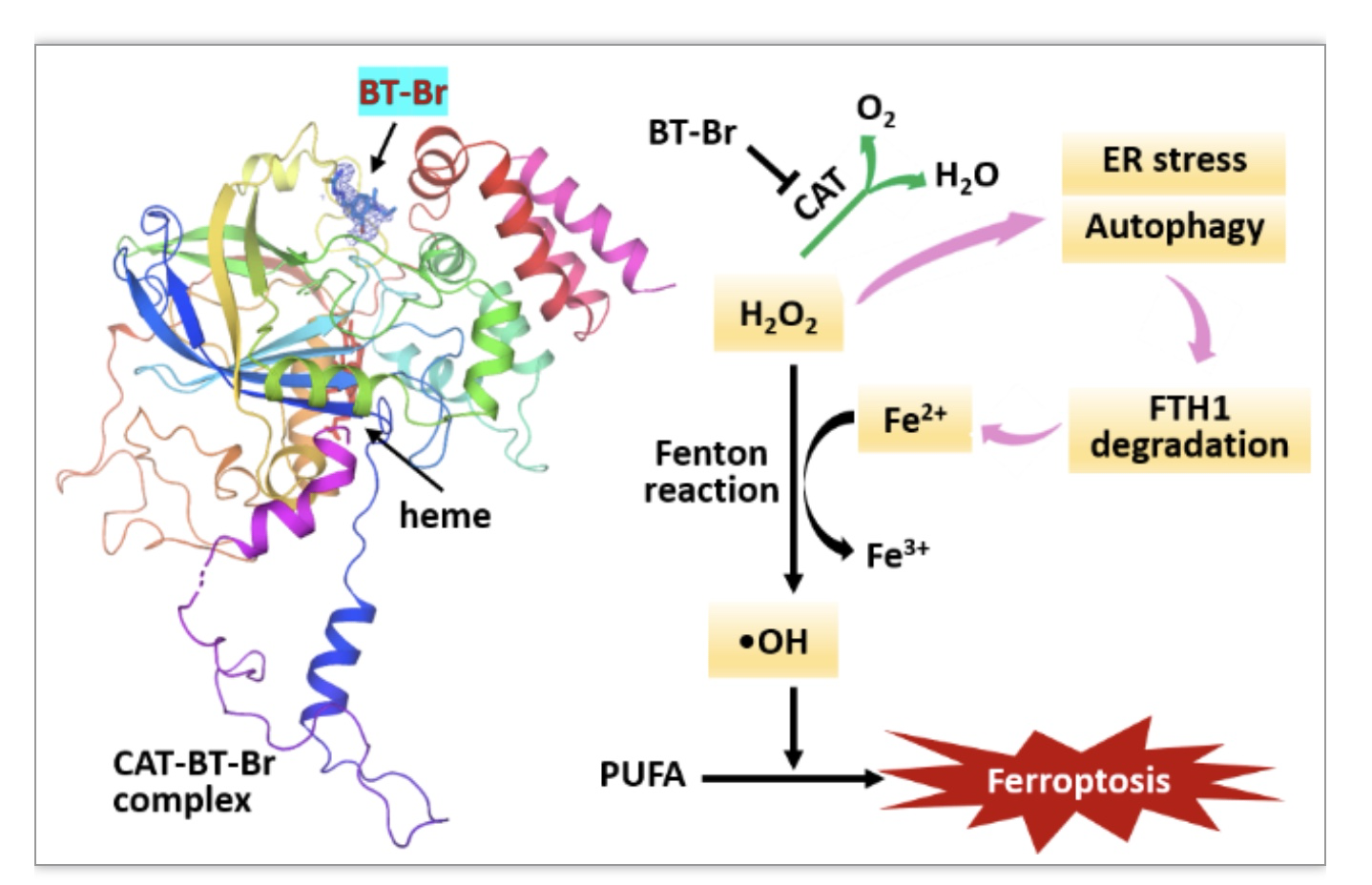
图1. (左)CAT-BT-Br的共晶结构(PDB ID:8HID)。(右)BT-Br诱导DU145细胞发生铁死亡的分子机制示意图。
