
全球每年超1000万人确诊急性肾损伤(acute kidney injury,AKI),造成约170万人死亡,严重威胁人民生命健康[1, 2]。临床手术造成的缺血再灌注、尿道梗阻等是诱导急性肾损伤的一个重要诱因[3];此外,铂类等化疗药物、硝苯地平等降血压药物、头孢类和氨基糖苷类抗生素、造影剂等多种药物使用亦可能诱发急性肾损伤[4]。急性肾损伤常引发胞外基质增多、肾小管萎缩、细胞死亡,导致肾功能障碍与肾脏不可逆损伤,诱发严重的慢性肾脏疾病CKD [5],最终发展成终末期肾病。急性肾损伤患者进展为CKD的风险为健康人群的8倍,全因死亡风险是健康人群的2倍[6]。急性肾损伤发病率高,死亡率高,严重威胁人类健康,但发病机制复杂,且尚无有效的药物治疗策略[7, 8],开展其关键治疗靶点研究,并据此设计治疗药物,具有重要的基础研究和临床转化意义。

肾损伤分子KIM1是一种跨膜糖蛋白,主要在肾脏中表达,生理状态下表达水平较低[9],但在急性肾损伤中,KIM1表达急速上调,可迅速、灵敏、特异地反映肾脏损伤程度,因此被FDA批准作为多种肾脏疾病的早期生物标志物[10]。此外,由于KIM1表达于上皮细胞,可介导多种胞内外信号传递,发挥多种效应,被报道参与病毒入侵、免疫应答、组织损伤与修复、肿瘤发生发展等多种生理病理活动中[11, 12]。KIM1分子在急性肾损伤发展过程中扮演什么样的角色?具体分子作用机制是什么?抑制KIM1与其下游效应分子的作用能否作为急性肾损伤的治疗手段?如何据此设计急性肾损伤治疗药物?这一系列重要科学问题目前仍有待深入研究。
KIM1结构简图如下所示(图1a)。研究人员发现, KIM1在AKI模型中显著上调(图1b-d)。KIM1过表达肾小管上皮细胞中,给予顺铂刺激,可促进顺铂导致的细胞凋亡和炎症因子上调;KIM1敲除则可缓解顺铂所诱导的细胞凋亡,并抑制炎症因子的表达(图1e-h)。进一步研究发现,KIM1过表达后,促进了顺铂所诱导的多聚ADP核糖聚合酶1(poly-ADP-ribose polymerase 1,PARP1)的剪切及p53的磷酸化,加剧凋亡;KIM1敲除则抑制顺铂所诱导的PARP1的剪切及p53的磷酸化(图1i-j)。上述结果共同表明KIM1影响顺铂诱导的细胞损伤可能是通过促进细胞凋亡实现的。

图1 KIM1在AKI后显著上调并加重炎症和凋亡反应
(图源:Chen Y, et al., Nat Commun, 2023)
Co-IP和FRET实验证明KIM1与DR5间存在结合,且在损伤条件下,KIM1与DR5的结合有增强趋势(图2a-d)。DR5在肾损伤后显著上调,且与KIM1有明显的共定位(图2e, f)。敲低DR5能抵抗KIM1加重的顺铂诱导的凋亡(图2g)。KIM1过表达会促进顺铂诱导的DR5寡聚化,表现为FRET信号的增强;而KIM1敲除则抑制顺铂诱导的DR5寡聚化,表现为FRET信号的减弱(图h, i)。非变性凝胶电泳的结果表明,顺铂刺激下,KIM1过表达促进DR5高分子量寡聚体(High-ordered oligomers)的形成,KIM1敲低则抑制DR5高分子量寡聚体的形成(图j, k)。进一步研究发现,KIM1可与DR5全长、DR5ΔTMH和DR5ΔCytD结合,而不与DR5ΔECD结合,表明ECD为DR5结合KIM1的关键区段(图2l)。同时,过表达KIM1相关截短体(Ig V,Mucin,TM+CytD)及DR5全长,发现KIM1 Ig V区段是其与DR5结合的关键区段。提示顺铂刺激下KIM1促进DR5寡聚化。
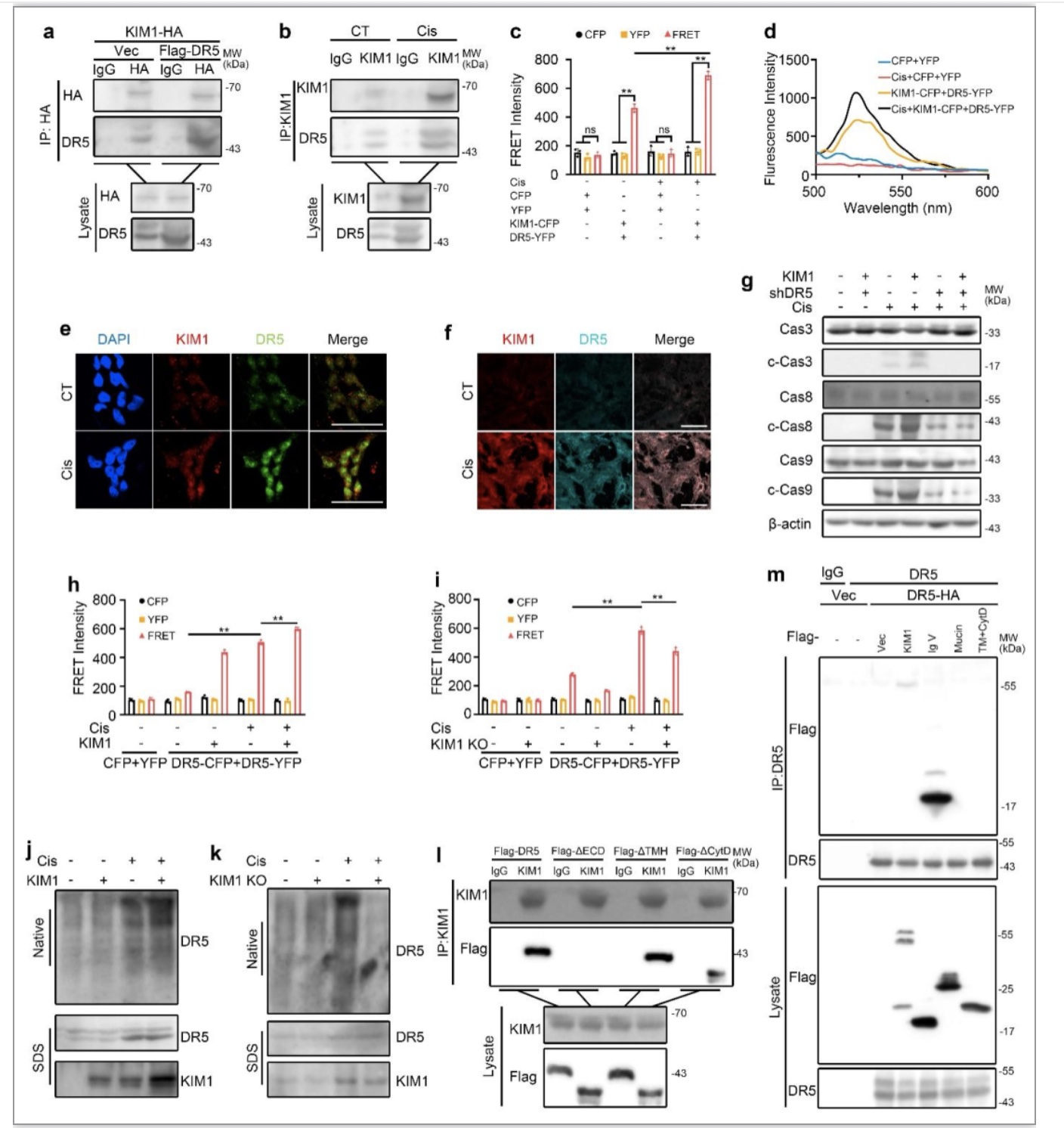
图2 KIM1结合DR5并促进其寡聚化
(图源:Chen Y, et al., Nat Commun, 2023)
顺铂诱导的AKI小鼠模型中, 肾小管特异性敲除Kim1(Kim1Ksp-KO)小鼠血清肌酐和尿素氮水平均低于WT小鼠,肾小管上皮细胞脱落及管腔扩张等现象减少,表明Kim1Ksp-KO小鼠的损伤较轻(图3a-c)。肾小管特异性敲除Kim1抑制了Caspase3,8,9的活化(图3d),表明DR5下游信号通路被抑制。Kim1Ksp-KO小鼠肾脏TUNEL阳性着色相较于WT小鼠显著减少(图3e),表明Kim1敲除在小鼠水平缓解了顺铂诱导的凋亡。同时,肾小管特异性敲除Kim1抑制了DR5高分子量寡聚体的形成(图3f)。此外,在bIRI诱导的AKI小鼠模型中得到了相同的结论,即肾小管特异性敲除Kim1通过抑制DR5下游凋亡信号缓解bIRI诱导的肾损伤(图3g-l)。这些结果说明肾小管特异性敲除Kim1缓解顺铂及缺血再灌注诱导的AKI。
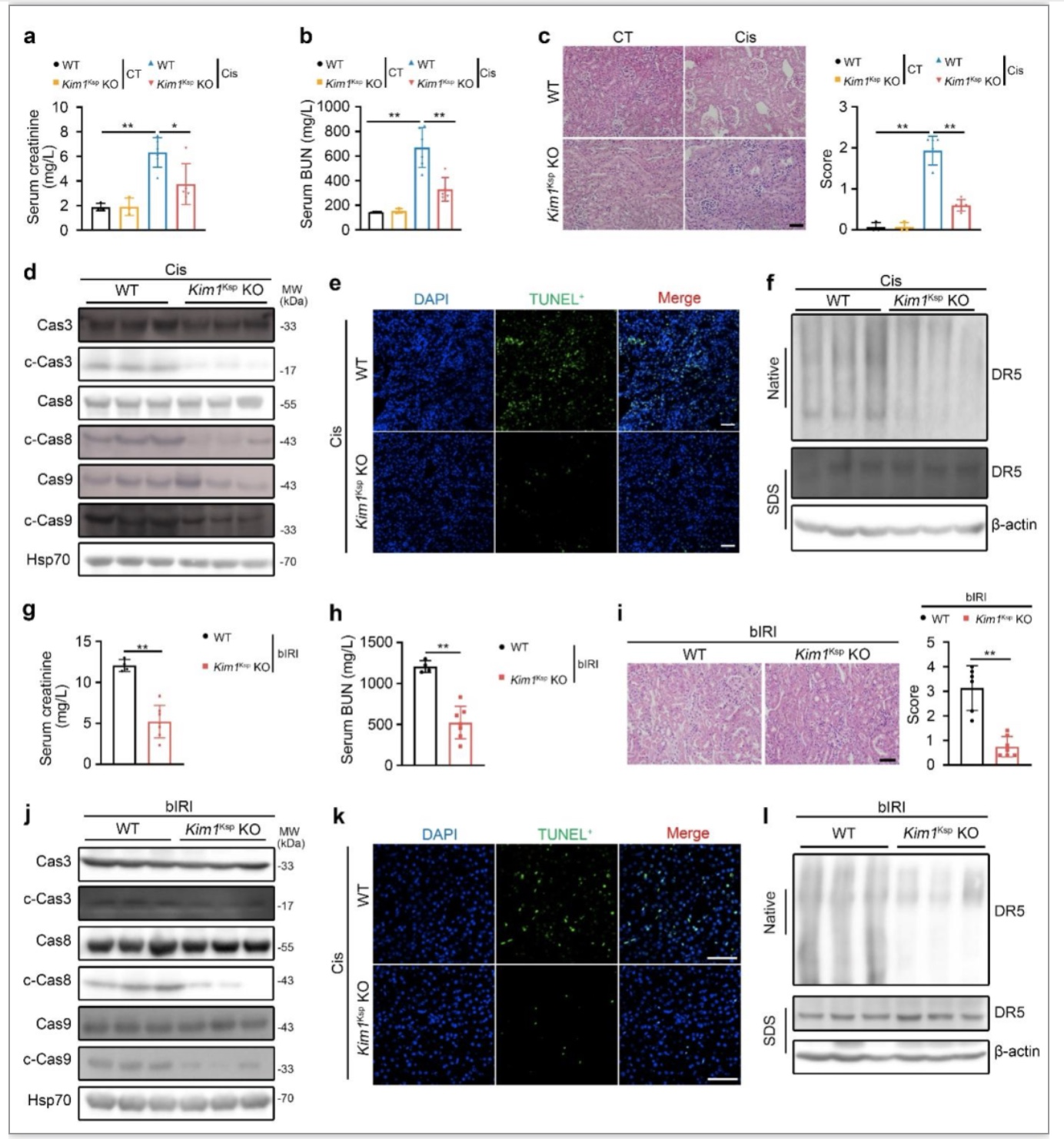
图3 肾小管特异性敲除Kim1缓解顺铂及缺血再灌注诱导的AKI
(图源:Chen Y, et al., Nat Commun, 2023)
针对KIM1与DR5的结合位点,利用AlphaFold2和Human KIM1-DR5 PPI进行拮抗肽筛选,结合细胞验证获得拮抗肽P2(图4a, b)。P2可抑制顺铂导致的损伤相关分子表达以及Caspase3,8,9的活化,促进抗凋亡分子的表达(图4c, d)。进一步研究证明, 5’(6) FAM标记的P2在顺铂诱导的AKI小鼠模型中与KIM1和DR5有良好的共定位(图4e)。在顺铂诱导的AKI动物模型中,P2尾静脉注射显著改善了肾功能,同时改善了肾脏病理损伤(图4f-h)。P2显著抑制了顺铂所诱导的Caspase3,8,9的激活,改善了顺铂所诱导的凋亡(图4i, j)。肾脏组织Co-IP的结果表明,顺铂刺激下,P2可阻滞KIM1与DR5的结合(图4k)。上述结果表明,顺铂损伤下P2可到达KIM1与DR5互作部位,阻滞KIM1与DR5的结合,从而抑制凋亡,改善AKI。
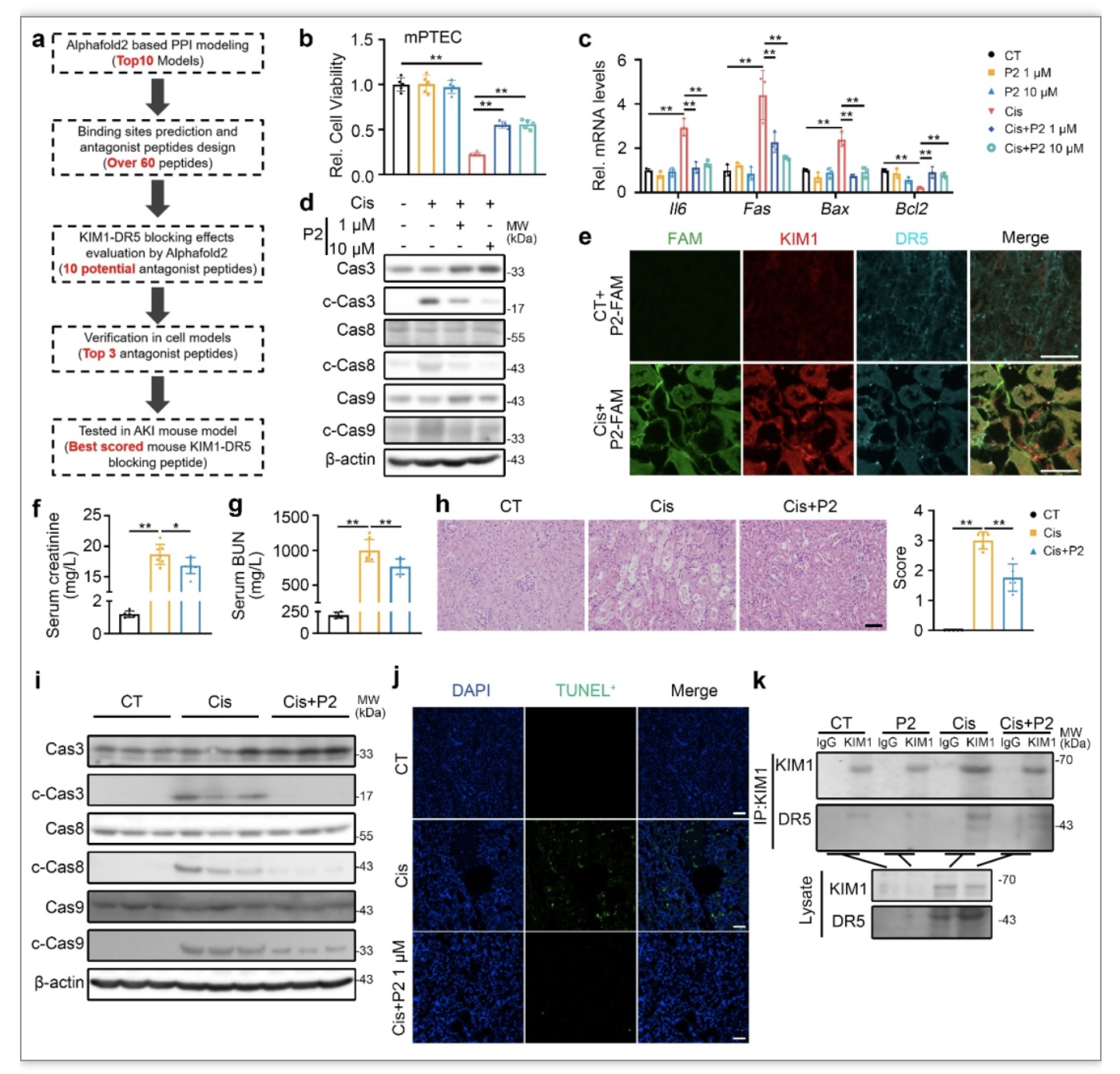
图4 拮抗肽P2干预KIM1-DR5互作缓解急性肾损伤
(图源:Chen Y, et al., Nat Commun, 2023)

图5 YY1-KIM1-DR5轴促进AKI发展及潜在治疗策略
(图源:Chen Y, et al., Nat Commun, 2023)
