

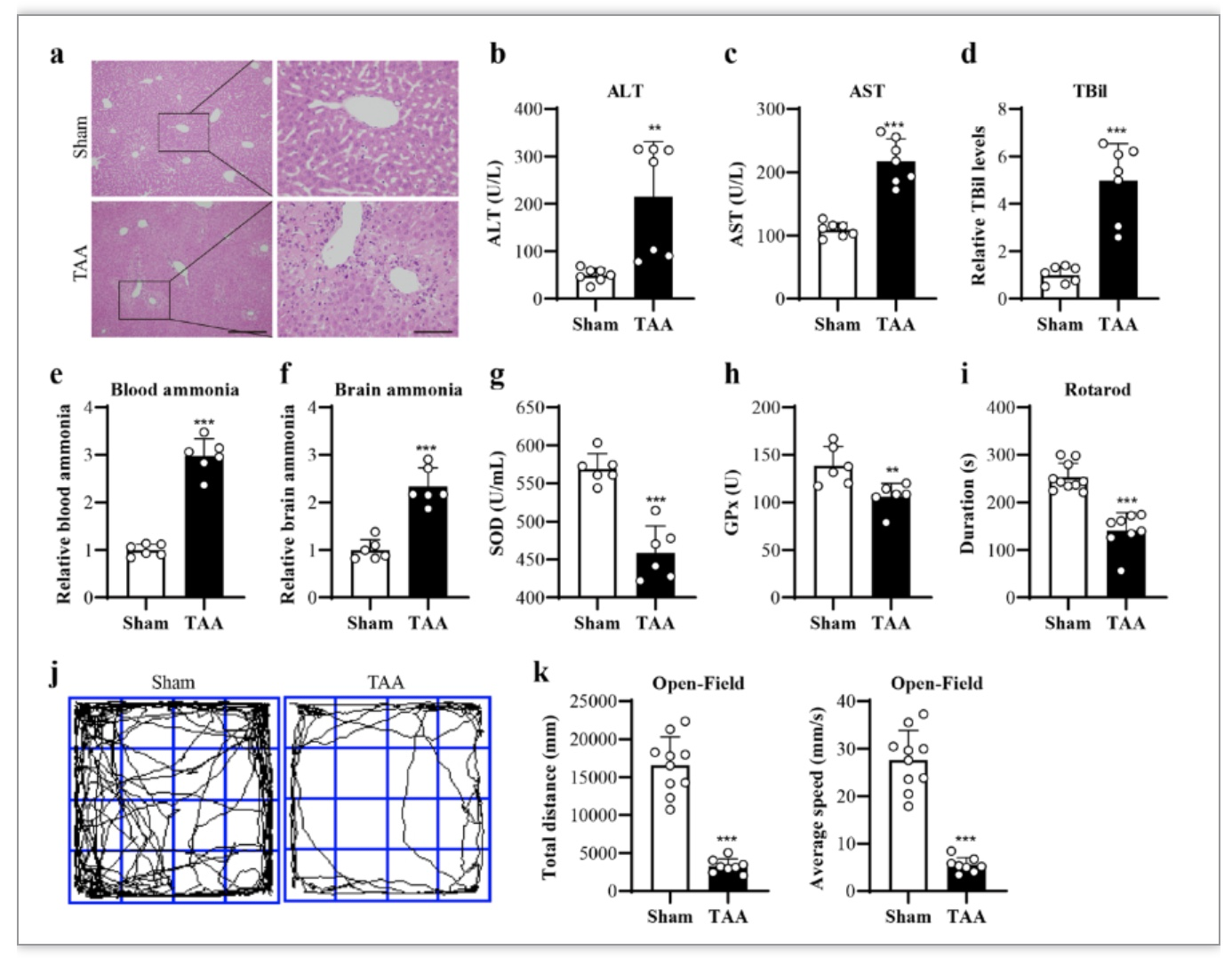
首先,将连续3天的150 mg/kg TAA注射用于研究。研究发现,TAA诱导了肝损伤,H&E染色显示肝细胞坏死(图1a),血ALT、AST和TBil水平升高(图1b–d),血氨(图1e),以及Elisa显示的脑氨(图1f)。此外,TAA诱导了全身氧化应激,表现为血液SOD和GPx水平显著降低(图1g,h),并受到抗氧化活性生物标志物的影响。通过转棒测试,TAA小鼠表现出平衡障碍(图1i和见下文),通过旷场测试,总距离和自发运动速度降低(图1j,k)。这些数据表明,TAA在小鼠中诱导了HE样症状和全身氧化应激。
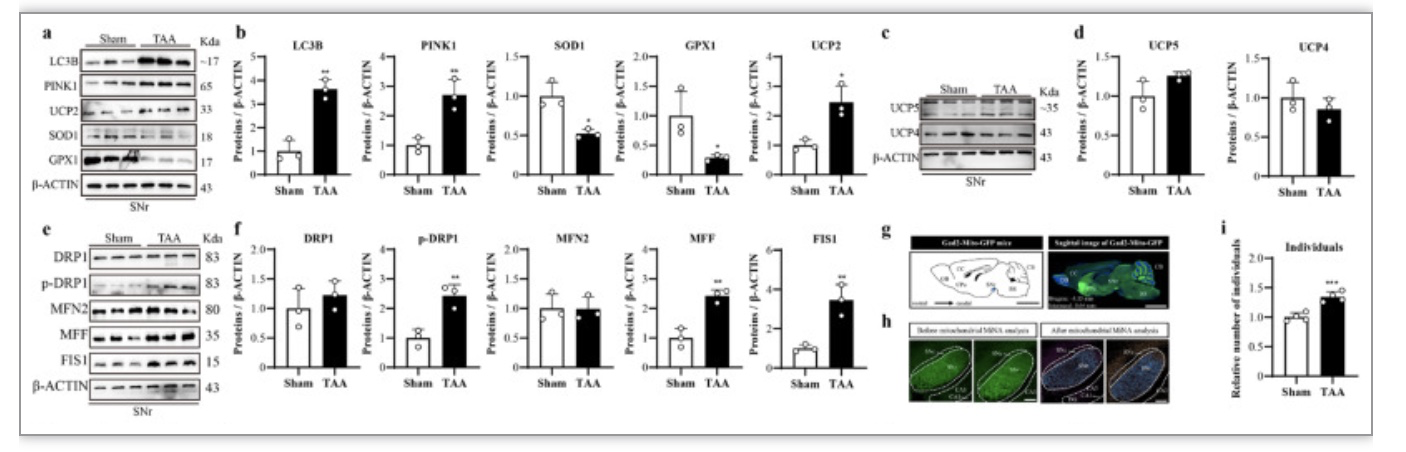
SNr在介导HE的运动缺陷中起着关键作用。大脑皮层是另一个受到高氨血症和氧化应激攻击的靶点。考虑到这一点,检测了自噬标记物(LC3B和PINK1)、抗氧化应激标记物(SOD1和GPx1)、线粒体解偶联蛋白(UCP2、UCP4和UCP5)和线粒体因子的表达水平,包括动力蛋白相关蛋白1的线粒体前分裂因子(DRP1)及其磷酸化版本的p-DRP1,在SNr和大脑皮层中,负责中央分裂的线粒体分裂因子(MFF)、负责周边分裂的线粒体分裂蛋白1(FIS1)和线粒体融合蛋白2(MFN2))。蛋白质印迹结果所示,在SNr中,TAA诱导自噬、线粒体分裂以及氧化应激指标均明显增加。
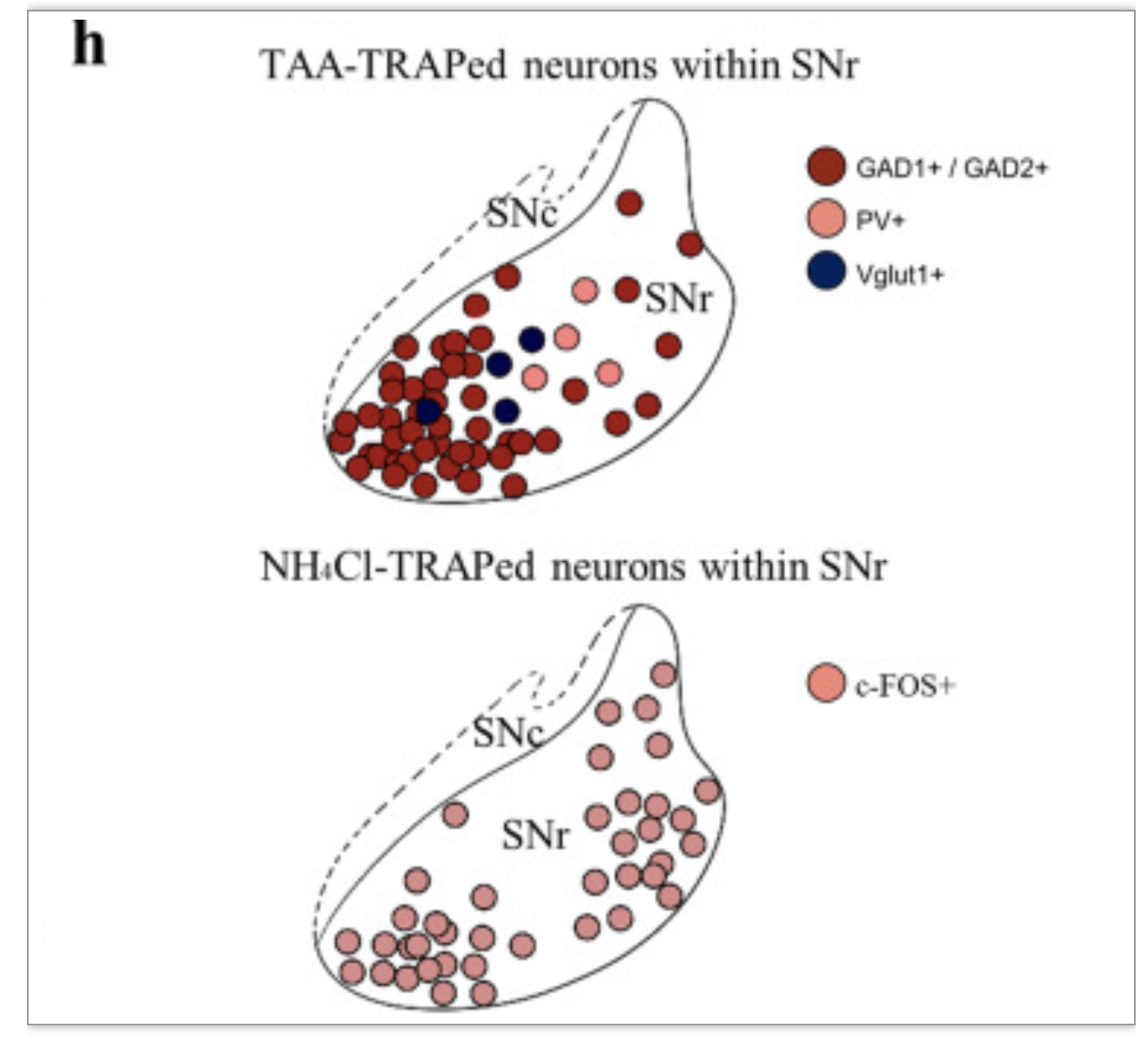
采用活性群体靶向重组(TRAP)方法选择对特定刺激激活的神经元的通路,以获得对HE诱导的神经元的遗传通路。在双侧SNr内注射pAAV-hSynDIO-mCherry病毒载体的FOS cre ERT2(TRAP2)小鼠用于靶向TRAPing至SNr区域,并将标记限制在神经元群体中。令人惊讶的是,mSNr观察到TAA-TRAPed细胞的急剧增加,而不是lSNr。此外,还分析了FOS+神经元最常表达的标志物类型。hSyn启动子的有效性在图中得到了证实。如图所示,90%的TRAPed细胞是GABA能(表达GAD1、GAD2或PV),其中大多数表达为GAD1或GAD2(81%),而不是PV(8%)。在此,TAA在mSNr处主要激活表达GAD2的GABA群体。
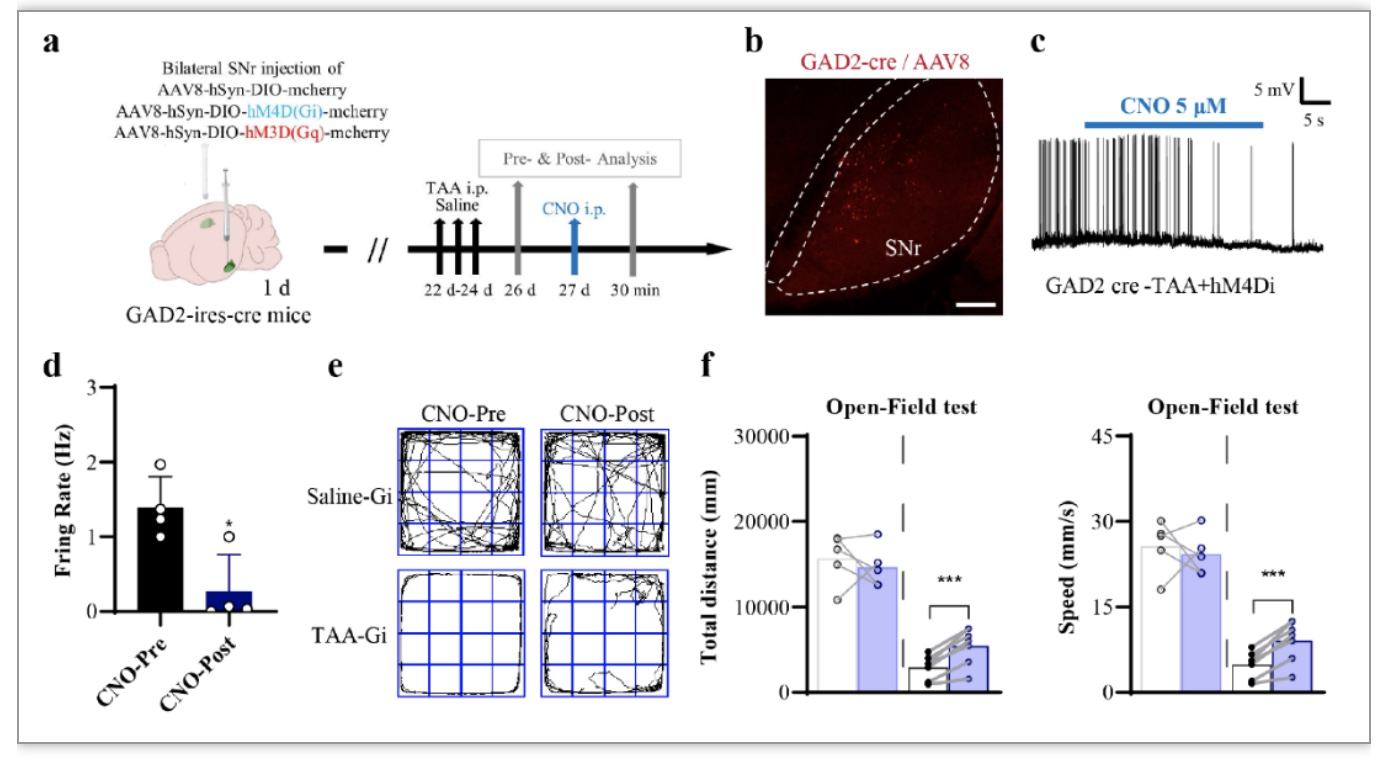
DREADD是一种基因修饰的毒蕈碱受体,对内源性乙酰胆碱没有亲和力,而受体可以被CNO(或DCZ)激活,CNO是非典型抗精神病药物氯氮平的一种药理学惰性代谢产物。DREADD可以通过Gq或Gi途径分别偶联以刺激或抑制神经元活动。使用两个配体(CNO和DCZ)来激活hM4D(Gi)受体和hM3D(Gq)受体,实验程序如图4a所示。。Gi或Gq病毒载体被注射用于化学遗传学操作。冠状切片上病毒载体的共焦照片证实了正确的立体定位(图4b)。膜片钳记录用于证明Gi组中表达红色mCherry的SNr神经元的电生理特征。结果证实了这些化学遗传学抑制的神经元的放电减少(图6c)。与CNO前组相比,CNO后组中表达GAD2的GABA群体的放电率急剧下降(图6d)。旷场试验的结果证实了Gi组的运动改善(图6e、f)。
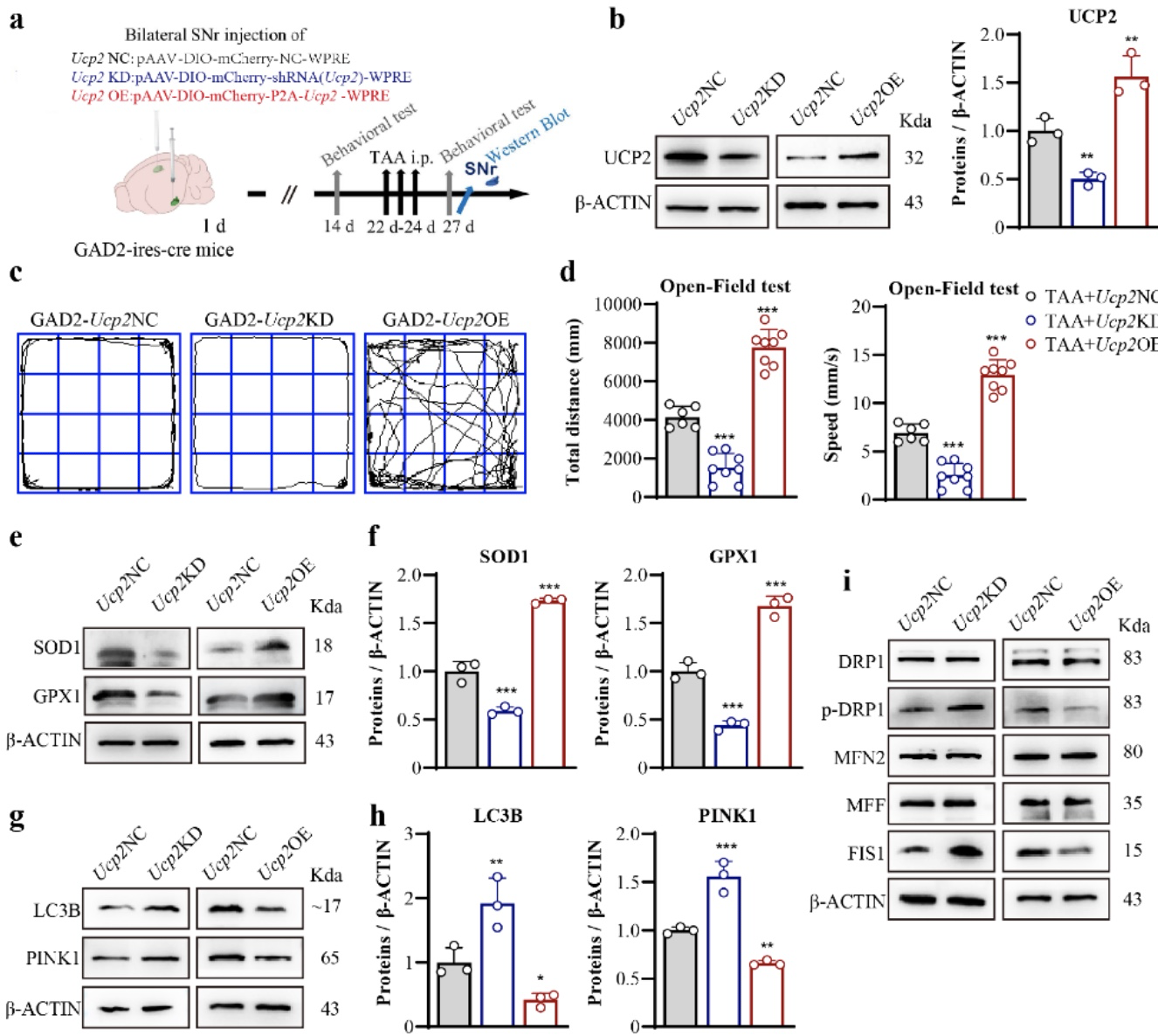
进一步,研究了UCP2通过调节氧化应激和线粒体动力学对SNrGAD2神经元HE行为的保护作用。作为一种线粒体蛋白,UCP2在线粒体功能中发挥着关键作用,包括将氧化磷酸化与ATP合成分离,并控制质子重新进入线粒体基质29。将UCP2病毒载体注射到GAD2 ires-cre小鼠的双侧SNr中,以选择性靶向表达GAD2的GABA群体(图5a段)。Ucp2-OE治疗减轻了TAA小鼠的运动迟缓,而Ucp2-KD治疗加重了运动迟缓(图5c,d)。研究表明,Ucp2-OE处理减轻了氧化应激(图5e,f)、自噬(图5g,h)和线粒体断裂(图5i,j)。相反,Ucp2-KD处理加重了氧化应激、自噬和线粒体断裂。这些结果表明,针对SNrGAD2群的Ucp2-OE改善了HE。
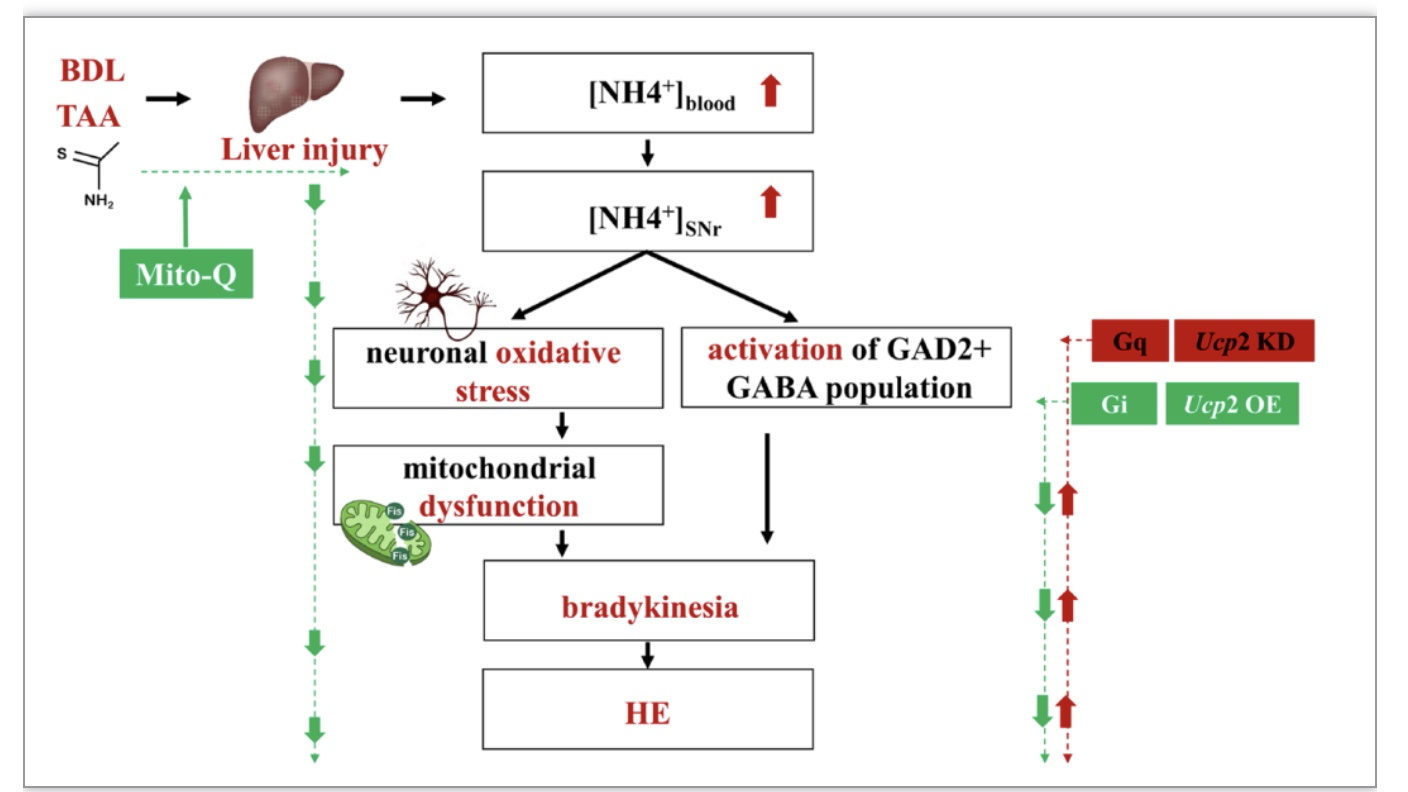
总之,该研究表明HE或局部高氨刺激可以直接激活内侧SNr区表达GAD2的GABA群体。此外,抑制该群体的化学遗传,或在该群体中靶向过表达线粒体UCP2,或系统性应用线粒体靶向抗氧化药物Mito-Q均可有效改善HE。该研究提示SNrGAD2群体线粒体对氨失衡敏感,可作为HE治疗的中心。本研究为线粒体与HE的关系提供了一个新的视角。
